Diagnosis and management of vascular Ehlers-Danlos syndrome: Experience of the UK national diagnostic service, Sheffield | European Journal of Human Genetics

Mendelian Genetics: Patterns Of Inheritance And Single-Gene Disorders
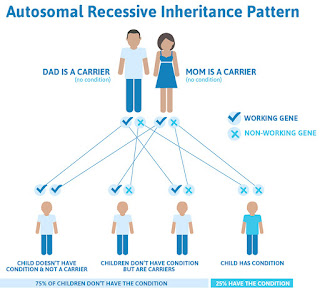
Autosomal recessive single-gene diseases occur only in individuals with two mutant alleles of the disease-associated gene. Remember, for any given gene, a person inherits one allele from his or her mother and one allele from his or her father. Therefore, individuals with an autosomal recessive single-gene disease inherit one mutant allele of the disease-associated gene from each of their parents. In pedigrees of families with multiple affected generations, autosomal recessive single-gene diseases often show a clear pattern in which the disease "skips" one or more generations.
Phenylketonuria (PKU) is a prominent example of a single-gene disease with an autosomal recessive inheritance pattern. PKU is associated with mutations in the gene that encodes the enzyme phenylalanine hydroxylase (PAH); when a person has these mutations, he or she cannot properly manufacture PAH, so he or she is subsequently unable to break down the amino acid phenylalanine, which is an essential building block of dietary proteins. As a result, individuals with PKU accumulate high levels of phenylalanine in their urine and blood, and this buildup eventually causes mental retardation and behavioral abnormalities.
The PKU-associated enzyme deficiency was determined biochemically in the 1950s—long before the PAH-encoding gene was mapped to human chromosome 12 and cloned in 1983. Specifically, Dr. Willard Centerwall, whose child was mentally handicapped, developed the first diagnostic test for PKU in 1957. Called the "wet diaper" test, Centerwall's test involved adding a drop of ferric chloride to a wet diaper; if the diaper turned green, the infant was diagnosed with PKU. The wet diaper test was used to reliably test infants at eight weeks after birth; by this time, however, infants who were affected by PKU had already often suffered irreversible brain damage.
Thus, in 1960, Dr. Robert Guthrie, whose niece suffered from PKU and whose son was also mentally handicapped, established a more sensitive method for detecting elevated phenylalanine levels in blood, which permitted a diagnosis of PKU within three days after birth. Guthrie's test used bacteria that were unable to make their own phenylalanine as messengers to report high blood levels of phenylalanine in an infant's blood sample obtained via heel prick. With Guthrie's method, the phenylalanine-deficient bacteria were grown in media together with a paper disk spotted with a drop of the infant's blood. If the phenylalanine levels in the blood were high, the bacteria would grow robustly, and a diagnosis of PKU could be made. Through the ability to discover that their child had PKU at such an early age, parents became able to respond immediately by feeding their child a modified diet low in proteins and phenylalanine, thereby allowing more normal cognitive development. Guthrie's test continues to be used today, and the practice of obtaining an infant's blood sample via heel prick is now used in numerous additional diagnostic tests.
Several other human diseases, including cystic fibrosis, sickle-cell anemia, and oculocutaneous albinism, also exhibit an autosomal recessive inheritance pattern. Cystic fibrosis is associated with recessive mutations in the CFTR gene, whereas sickle-cell anemia is associated with recessive mutations in the beta hemoglobin (HBB) gene. Interestingly, although individuals homozygous for the mutant HBB gene suffer from sickle-cell anemia, heterozygous carriers are resistant to malaria. This fact explains the higher frequency of sickle-cell anemia in today's African Americans, who are descendants of a group that had an advantage against endemic malaria if they carried HBB mutations. Finally, oculocutaneous albinism is associated with autosomal recessive mutations in the OCA2 gene. This gene is involved in biosynthesis of the pigment melanin, which gives color to a person's hair, skin, and eyes.
X-chromosome Study Reveals Hidden Genetic Links To Alzheimer's Disease
Despite decades of research, the X-chromosome's impact on Alzheimer's was largely ignored until now. Explore how seven newly discovered genetic loci could revolutionize our understanding of the disease.
Study: X‐chromosome-wide association study for Alzheimer's disease. Image Credit: nobeastsofierce / Shutterstock
Conventional investigations of the genetic contributors to Alzheimer's disease (AD) risk and progression have ignored the role of the X-chromosome, primarily due to technical analysis limitations. To address these knowledge gaps, a recent study published in the journal Molecular Psychiatry leveraged extensive X-Chromosome-Wide Association Study (XWAS) data from 115,841 AD cases (including clinically diagnosed and proxy cases) and 613,671 controls to identify genetic signals indicative of AD pathophysiology.
The study considered three patterns of X-chromosome inactivation (XCI) in females (r-XCI, s-XCI, and e-XCI) and found no AD-associated genome-wide signals in the non-pseudoautosomal regions of the X-chromosome. Notably, the study identified seven loci with X-chromosome-wide significance thresholds that may contribute to AD-associated genes (e.G., FRMPD4, DMD, and WNK3), which were highlighted as essential targets for future research.
BackgroundAlzheimer's disease (AD) is an age-associated neurodegenerative disorder characterized by progressive memory and cognitive decline. It remains the most common precursor to adult dementia, with hitherto no identified cure. Decades of research have highlighted several (>80) genetic contributors (loci) to AD risk. Unfortunately, traditional technical limitations have resulted in the X-chromosome being predominantly excluded from these investigations.
The X-chromosome comprises 5% of the genome, with previous research suggesting it contains up to 15% of known genetic intellectual disability-contributing genes. Significant sexual dimorphism (male versus female differences) in both X-chromosome properties (women have two X-chromosomes, while men have only one) and AD outcomes (women are at higher AD risk and live longer with AD than their male counterparts, while men demonstrate more rapid AD-associated cognitive decline) necessitates enhanced understanding of the X-chromosome's role in AD risk and progression.
About the StudyThe present study aimed to address gaps in our understanding of the X-chromosome's role in AD risk and progression by using an in-depth X-Chromosome-Wide Association Study (XWAS). The study dataset was derived from 35 previous studies, two independent family cohorts, and two biobanks (UK Biobank [UKB] and FinnGen). It included 115,841 AD cases (52,214 clinically diagnosed and 55,868 proxy cases), AD-proxies (defined as 'either parent demonstrating dementia' in females, and 'mothers demonstrating dementia' in males), and 613,671 controls (55% women), all of whom were of European ancestry.
a Main analyses and b sensitivity analyses. Box colors indicate the approach: purple, green, orange and blue represent r-XCI, s-XCI, e-XCI and sex-stratified approaches, respectively. Boxes circled in red are the main r-XCI, s-XCI and e-XCI analyses. *Fixed effect meta-analysis with an inverse-variance weighted approach as implemented in METAL. **Sex-stratified models were adjusted on 1) principal components (PCs) and/or the genotyping center; 2) PCs, center and age; 3) PCs, center, age and APOE.
Following sensitivity analyses, 63,838 diagnosed AD cases and 806,335 controls were included for downstream analyses. The study further incorporated cerebrospinal fluid biomarker analyses (Aβ42 and pTau) and cognitive impairment assessments (Mini-Mental State Examination [MMSE]) in a subset of included participants (5,522 and 2,661, respectively). Notably, the study excluded pseudoautosomal regions from the analyses, primarily due to their exclusion from most participants' genotyping chips.
Analytical computation included association tests carried out under three X-chromosome inactivation (XCI) regimes accounting for different female XCI states – 1. Random XCI (r-XCI), 2. Skewed XCI (s-XCI), and 3. Escape XCI (e-XCI). Researchers additionally conducted sex-stratified analyses to account for variability induced by XCI mechanisms, which could result in stronger-than-expected effect sizes in males. Stringent quality control measures and sensitivity analyses were applied to ensure high data reliability and to mitigate potential false negatives arising from biobank-specific methodological differences.
"To maintain balance around allelic dosage between the sexes, X-chromosome inactivation (XCI) occurs in females. This process is where one X chromosome is transcriptionally silenced during female development. The choice of the silenced copy is most often random (random XCI or r-XCI), but inactivation can also be skewed toward a specific copy (skewed XCI or s-XCI). Importantly, up to one‐third of X‐chromosome genes 'escape' inactivation and are expressed from both X‐chromosomes in female cells (escape XCI or e-XCI)."
Finally, genetic colocalization computations comparing study outcomes (identified genetic loci) with preexisting protein- and expression-quantitative trait loci (pQTL and eQTL, respectively) datasets were employed to identify traits and biomarkers representative of cognitive decline.
Study FindingsThe XWAS analyses conducted herein identified 666,264 r-XCI, 442,001 e-XCI, and 438,420 s-XCI variants, of which 288,320, 276,902, and 263,169, respectively, were common (minor allele frequency [MAF] ≥ 1%). Notably, none of the approaches employed identified genome-wide significant signals, suggesting that the non-pseudoautosomal regions of the X-chromosome are devoid of common AD-associated genetic risk factors.
Seven loci with X-chromosome-wide significance thresholds were identified, including four common loci (Xp22.32, FRMPD4, DMD, and Xq25) and three rare loci (WNK3, PJA1, and DACH2). These loci are highlighted as targets for future investigation and may hold the key to discovering clinical, therapeutic, and pharmacological interventions against AD genesis and progression.
FRMPD4, a brain-expressed gene linked to cognitive reserve, showed particularly robust signals. In contrast, rarer variants such as those in PJA1 and DACH2 demonstrated poor data quality (e.G., sparse variant coverage and lower imputation quality), underscoring the need for methodological optimizations in future research.
ConclusionsThe present study represents the largest XWAS on AD to date, analyzing data from over 115,000 cases and 613,000 controls. It presents the first attempt at accounting for X-chromosome complexities, such as variability in female XCI patterns and the limitations of biobank-specific methods. While no genome-wide significant associations were found, seven suggestive loci, including FRMPD4, DMD, and WNK3, were identified. In tandem with gene expression and epigenetic investigations, this study may form the basis of future clinical interventions against AD risk and progression.
Journal reference:


Comments
Post a Comment