Coagulation disorders

Examining The Dark Part Of The Genome To Find Developmental Causes For Facial Disorders
Elizabeth Engle, MD, has devoted her career to finding genetic and developmental causes for disorders of eye, eyelid, and facial movement. From common conditions like strabismus to very rare disorders, these conditions can impact a person's appearance and impair social communication, making it hard to shift one's eyes up, down, or sideways or adjust facial expressions.
Each discovery in Engle's lab offers an answer for families. It's also a window into the brain—specifically into the development of the nerves and motor neurons that innervate the eye and face muscles.
Now, after the better part of a decade, Engle's team in the F.M. Kirby Neurobiology Center at Boston Children's Hospital drove the cracking of a previously impenetrable mystery: the genetic cause of a condition called hereditary congenital facial paresis type 1 (HCFP1). The study is published in the journal Nature Genetics.
HCFP1 causes mild facial weakness that makes it hard for babies and children to smile brightly, pucker their lips, suck, and sip from straws. While it's mild and very rare, the condition has opened the door to a little-known area of genetics: disorders caused by inherited mutations in the genome's "dark matter."
A rare category of genetic diseaseMost known genetic causes of inherited disease come from variants in genes—the 1–2% of our genome, also known as the exome, that provides the code for making proteins. But within the rest of the genome—the dark matter—lie important noncoding elements that regulate genes and can influence disease.
"While we only have about 20,000 genes, noncoding regulatory elements provide much additional information, modulating gene expression by turning genes on or off in specific cell types at specific times," says Engle.
Inherited, single-gene disorders involving non-coding variation remain mostly under-explored, in part because we don't yet know how to interpret most variation within the vast non-coding space. The new work provides a rare example that could inform future genetic discovery for other disorders.
Examining families' genetic variantsThe team started with nine families from the Engle lab's database and five families from collaborators in the Netherlands. Linkage mapping in the Dutch families had identified the general area in the genome that seemed tied to HCFP1. The families had had whole-exome sequencing, but since this includes only the coding parts of the genome, it came up empty.
Engle's team used whole genome sequencing, which revealed that five families had a large, duplicated piece of DNA. The start and end points of this piece varied, but in all five families it contained three non-coding regulatory elements. The other nine families had single nucleotide variants (SNVs)—single "letter" changes in the genetic code—all within one of the three regulatory elements.
"That's a bit of a conundrum, because we typically think of DNA duplications as increasing gene expression, and SNVs as decreasing gene function," says Engle. "We had to figure out the mechanism by which these duplications and SNVs resulted in the same facial phenotype and determine what gene these regulatory elements were targeting."
They found that the three regulators are located close to GATA2, a gene known to help specify the production of different cell types in the blood and nervous system. That raised the question: Do the three regulators act on GATA2? Engle's team suspected they did, and that two enhanced and one silenced GATA2 expression.
Tracking cells' developmental journeysThat's where much legwork came in. Using mouse models and tools like single-cell RNA sequencing and CUT&Tag to profile cells at different time points, the team began to understand how variants in DNA regulatory elements lead to facial weakness.
First, they found that the regulatory elements are active during early brain development, in a location in the hindbrain where two cell types are born: facial motor neurons and inner ear efferent neurons (involved in filtering loud sounds and background noise).
"Our hypothesis was that inner ear efferents are born first and are dependent on the enhancers turning on GATA2, and that a day or so later, the silencer shuts down GATA2 and facial motor neurons are made," says Engle. "We thought that the DNA duplications could increase enhancer activity while the SNVs could reduce silencing activity—both of which would lead to production of more inner ear efferents and fewer facial motor neurons."
Meticulous experiments showed this to be true. "The combined number of cells was unchanged, but prolonging GATA2 expression increased the inner ear efferents at the expense of the facial motor neurons," says Alan Tenney, Ph.D., one of the postdoctoral research fellows in Engle's lab who led the study.
An explanation for HCFP1In short, the team showed that the genetic mutations prevent people from making enough motor neurons during development to properly innervate the facial muscles, leaving them relatively weak.
"To solve questions like this, one needs a team of people with multiple complementary areas of expertise," says Engle. "We think our work provides an example of how to tease out the genetics and mechanisms of inherited disorders involving non-coding variation. The key is what you choose to tackle—which non-coding variants are compelling enough to spend the next five years of your life investigating."
More information: Alan P. Tenney et al, Noncoding variants alter GATA2 expression in rhombomere 4 motor neurons and cause dominant hereditary congenital facial paresis, Nature Genetics (2023). DOI: 10.1038/s41588-023-01424-9
Citation: Examining the dark part of the genome to find developmental causes for facial disorders (2023, July 21) retrieved 21 July 2023 from https://medicalxpress.Com/news/2023-07-dark-genome-developmental-facial-disorders.Html
This document is subject to copyright. Apart from any fair dealing for the purpose of private study or research, no part may be reproduced without the written permission. The content is provided for information purposes only.
Fragile, Not Broken: Local Families Raise Awareness For Fragile X Syndrome
Nearly a year and a half ago, I penned my final "From the Editor" column for Lowcountry Parent, sharing publicly for the first time about my son Archer's diagnosis of Fragile X syndrome at 3 years old. A few months later, we learned my daughter Lorelei, who many of you have seen grow up through the pages of the magazine and our social media accounts, shared the same genetic condition.
Their diagnoses have been life-altering for our entire family, yet oddly a relief at the same time — a sentiment I've come to realize is not uncommon among families like ours.
"Our son Saylor was 13 years old when he was diagnosed," said Steve Lowry of Mount Pleasant. "We had been to every specialist and therapy you can imagine, but it seemed like each specialist we visited would say something different. While they were trying to do the best they could, none of the potential diagnoses ever seemed to fit."
"When we first started pursuing answers, there was only one pediatric psychologist in the Lowcountry at the time," added his wife Cara. "No one ever said, hey, have you thought about genetic testing?"
Steve and Cara Lowry of Mount Pleasant have two children with Fragile X Syndrome, Saylor, 22, and Victoria, 19. Both will be attending post-secondary education programs this fall.
ProvidedIt wasn't until nearly a decade later, during a pediatrician appointment, that a resident who had been working with geneticists at MUSC made the suggestion which ultimately led to discovering that not only Saylor, but also his younger sister, Victoria, had Fragile X syndrome.
"I saw him struggle all those years, and we knew something was going but we just didn't know what," said Cara.
The genetic testing results were like finding the missing piece to a puzzle.
What is Fragile X?
If you've never heard of Fragile X before, you aren't alone. I hadn't either until that day in July 2021 when I found myself sitting in Greenwood Genetic Center's Charleston office.
Those who are older may have heard of Martin-Bell Syndrome, the original term for Fragile X which dates back to 1943. It wasn't until the 1990s when genetic testing technologies improved and the FMR1 gene was discovered, making Fragile X a fairly "new" condition.
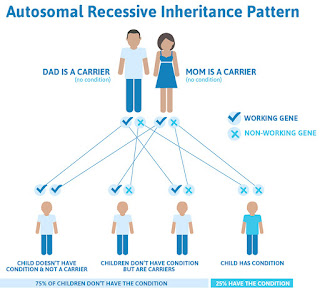
Fragile X is unique in that it is both a genetic and hereditary condition. This infographic from the National Fragile X Foundation shows how the Fragile X gene mutation is passed down.
According to the National Fragile X Foundation, Fragile X is a group of conditions associated with alterations in the FMR1 gene on the X chromosome — a gene responsible for the production of a protein called FMRP that is needed for brain development.
Genetic testing revealed I have a "premutation," which can lead to Fragile X-associated conditions and disorders like Fragile X-associated tremor/ataxia syndrome (FXTAS) or Fragile X-associated primary ovarian insufficiency (FXPOI). This premutation expanded to "full mutation" in both of my kids, causing their Fragile X syndrome.
It is estimated that about 100,000 Americans are living with Fragile X syndrome, classifying the condition as a rare disease. Rare diseases are defined as those with less than 200,000 individuals in the United States.
However, the Fragile X premutation is not rare. It's estimated that up to 1 in 151 individuals have the Fragile X premutation — many of whom are unaware until experiencing infertility or they have a child diagnosed with the full mutation.
"In both children and adults, Fragile X has a lot of characteristics in common with other conditions. For example, it's not uncommon for Fragile X-associated tremor/ataxia syndrome to be misdiagnosed as Parkinson's Disease," said Cara Lowry. "This is because many similar conditions are diagnosed via battery assessments versus a blood test. But we've learned just how important genetic testing can be. The results can impact the generation now, the generation to come, and the prior generation."
Fragile X and Autism
The full mutation, Fragile X syndrome, is the most prevalent inherited cause of mild to severe intellectual disability and the most common single-gene cause of autism spectrum disorder (ASD), accounting for about 1-6% of all cases of ASD.
It's important to mention the connection because there is a notable overlap in traits between the two that can sometimes lead to those with Fragile X Syndrome being misdiagnosed or co-diagnosed with autism.
In my family's case, Archer was first diagnosed with autism before receiving his genetic testing results. The Fragile X traits he displays are what cause him to meet the clinical criteria for ASD.
Research indicates about 15% of children with autism have been identified as having a genetic disorder, such as Fragile X syndrome, tuberous sclerosis, Down syndrome, or other chromosomal abnormalities, copy number variants, and single-gene mutations.
Defying the Odds
Like autism, individuals with Fragile X syndrome present with a spectrum of traits and characteristics. Having only one X chromosome, males tend to be more frequently and severely affected than females.
Now 5 years old, Archer is non-verbal, not potty trained and participates in occupational therapy, speech therapy, and music therapy to help manage his symptoms, promote communication and foster independence. His sister Lorelei, 8, showed no signs of developmental delay as an infant or toddler, and likely would have gone undiagnosed had it not been for him.
The Lowry's son, Saylor, who is now 22 years old, functions at a higher level of independence than many others living with Fragile X Syndrome. He earned his high school diploma, has completed an internship, is employed part-time and will soon be moving to attend the ClemsonLIFE program — a post-secondary education program with the specific purpose of helping young adults with intellectual disabilities obtain the life skills necessary to gain employment and live independently. Victoria Lowry, 19, will be attending Coastal Carolina University this fall.
The Lowrys will soon have an "empty nest," a milestone all parents anticipate but one they weren't sure would ever be their reality, as statistically only an estimated 10% of men with Fragile X and 40% of women are able to live independently.
"Like all parents, our goal has always been for them to be happy and live independently," said Steve.
Raising Awareness
On Friday, July 21, the Lowrys will join other parents, including myself, as well as families, self-advocates, state officials and researchers at the South Carolina State House to bring awareness to Fragile X syndrome and Fragile X associated disorders.
The ceremony is scheduled to take place one day before World Fragile X Day on July 22, which Governor Henry McMaster has also declared Fragile X Awareness Day in South Carolina.
In recognition, two prominent Lowcountry landmarks — Joseph P. Riley Jr. Ballpark and the Ashley River Bridge — will join more than 500 landmarks and monuments across 19 countries in "shining a light" on Fragile X by illuminating their locations the color teal.
To learn more about Fragile X, visit fragilex.Org or worldfragilexday.Com.
DHX9 Variations Underly Wide Spectrum Of Human Neurodevelopmental Disorders
A group of 20 patients with undiagnosed neurodevelopmental disorders ranging from severe to mild has now received a genetic diagnosis thanks to an international team of researchers at the GREGoR Research Center at Baylor College of Medicine, the Chinese University of Hong Kong, the German Mouse Clinic and collaborating institutions.
The team analyzed the patients' genes and conducted family studies to detect genetic mutations related to their condition. They discovered that the patients had mutations of the gene DHX9, which disrupted the gene's normal function. This is the first time that this gene has been associated with a human disease. Studies in animal models showed a connection between defective variations of the gene and neurodevelopmental problems. Altogether, the findings support that those variations of DHX9 underlie human neurodevelopment disorders and neuropathy. The study appears in the American Journal of Human Genetics.
"Our study started with two patients with remarkably different neurologic conditions for which they did not have a diagnosis despite extensive testing. Looking to find an answer to explain their condition, the patients joined our working group studying the genetic underpinnings or genomics of rare diseases," said first author Dr. Daniel Calame, instructor of pediatric neurology and developmental neurosciences and part of the GREGoR Research Center at Baylor. "In the beginning, we did not have any reason to believe that these patients had a genetic diagnosis in common. It was after we analyzed the results of their genome sequencing that we realized that each had a distinct unusual variant of gene DHX9. This motivated us to expand our efforts to find more cases, ultimately the 20 we came upon."
One of the surprising aspects of this study is that the patients' conditions are remarkably diverse. "Some patients have the most severe developmental disorders, including intellectual disability, seizures and movement disorders. Other patients have less severe conditions, for instance autism with normal IQ, while other patients have milder conditions – normal development but nerve degeneration leading to neuropathy, a condition typically causing numbness or weakness in adolescence or adulthood," Calame explained.
To begin to understand how variations in gene DHX9 can disturb neurodevelopment in such a variety of ways, the researchers conducted laboratory experiments in which the different DHX9 variants found in patients were introduced into cells and their functions compared to that of the DHX9 variant not associated with a condition.
"These cellular studies allowed us to distinguish quite clearly the functional alterations in the variants in the severe cases from those associated with the less severe or the mildest cases," said co-corresponding author Dr. Shen Gu, assistant professor in the School of Biomedical Sciences at the Chinese University of Hong Kong.
For instance, some variants associated with severe neurodevelopmental disorders were not located in the cell nucleus where the normal variant is typically located, but outside the nucleus in the surrounding cytoplasm.
"Another variant linked to severe neurodevelopmental disorders did not affect DHX9 localization but instead increased double-stranded DNA breaks, a process that negatively affects the integrity of the DNA and can disrupt the normal function of the cell," Shen said.
In addition to cellular studies, the researchers explored the effect of eliminating the Dhx9 gene in animal models. "Without the Dhx9 gene, the animals were less active in new environments and had a reduced sense of hearing, indicating a connection between the gene and neural functions," Shen said. "Our study shows DHX9 is involved in regulating mammalian neurodevelopment and neuronal well-being."
"This is an amazing story of international collaborative science, the global nature of human genetics research and the insights that can be gleaned by the study of neurological disease from a gene and genomic variation viewpoint – and achievements possible by the joining of scientific forces from two of my favorite cities: Hong Kong and Houston," said Dr. James R. Lupski, the Cullen Foundation Endowed Chair in Molecular Genetics, professor of pediatrics and molecular and human genetics and member of the Dan L Duncan Comprehensive Cancer Center at Baylor. Lupski also is co-corresponding author of this work and a principal investigator at GREGoR Research Center.
Find a complete list of the contributors to this work, their affiliations and financial support in the publication.


Comments
Post a Comment