Von Willebrand disease

Abnormal Uterine Bleeding May Be Caused By Clotting Factor Deficiencies
A new review article illuminates why physicians should keep coagulation disorders in mind when evaluating patients with heavy abnormal uterine bleeding.
Physicians treating patients with heavy menstrual bleeding (HMB) should consider the possibility that rare bleeding disorders and hemophilia carrier states are at play if other, more common causes of bleeding have been excluded, according to a new report.
The report, a literature review published in Life (Basel),1 outlines several coagulation disorders and offers physicians guidelines on how to spot and treat hemostatic disorders.
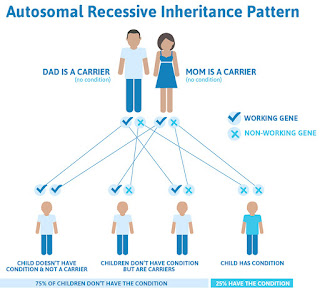
Authors explain that current literature suggests 10% to 35% of women experience HMB at some point during their reproductive years. There are several potential causes of such bleeding, and physicians can use the PALM-COEIN system to help identify the underlying cause.
This system of abnormal uterine bleeding classification, from the International Federation of Gynaecology and Obstetrics,2 includes structural causes like polyps, adenomyosis, leiomyoma, malignancy, and hyperplasia, and functional causes, including coagulopathy, ovulatory disorder, endometrial disorders, iatrogenic conditions, or not-yet-classified causes. The existing literature suggests that an underlying inherited bleeding disorder is at play for 10% to 62% of adolescents with HMB. The most common such disorder is von Willebrand disease, but the authors said several other, less-common conditions should also be considered. Those less-common conditions are the focus of the review article.
The authors first reviewed several rare coagulation factor disorders. For instance, they said factor I, or fibrinogen, is a soluble plasma glycoprotein that in some patients is absent (congenital afibrinogenemia) or reduced (hypofibrinogenemia). In the former, patients can have a wide range of clinical manifestations, from minimal bleeding to serious hemorrhages, the authors said. Such patients may also experience arterial or venous thrombosis. In coagulation tests, partial thromboplastin time (PTT), prothrombin time (PT), thrombin time (TT) are all "infinitely prolonged," the authors said, since all of the parameters require the formation of fibrin.
Patients with hypofibrinogenemia are often asymptomatic if their fibrinogen levels are above 1 g/L, but the authors said these patients sometimes experience bleeding or thrombotic complications.
"In affected individuals, PT, PTT, and TT are variably prolonged, with TT being the most sensitive assay," they said.
Factor VII deficiency is believed to make up about one-third of rare coagulation disorders. The factor is vitamin K dependent and, like factor I, is synthesized in the liver and secreted in the plasma, the authors said. Patients with a deficiency of factor VII can have a wide range of manifestations, the authors said, "from asymptomatic or mildly symptomatic cases—for example, easy bruising, gum bleeding, and epistaxis—to severe cerebral and gastrointestinal hemorrhage."
Unfortunately, there are no clear treatment guidelines for factor VII deficiency, the authors said, although the published literature suggests a number of potential treatments, including surgery and blood transfusions.
Factor XIII deficiency is one of the rarest coagulation disorders, with only 500 cases reported globally since 1960, the authors said.
"Factor XIII is a transglutaminase that cross-links fibrin fibers between amino acid residues, hence stabilizing a fibrin clot," they wrote.
Patients with the deficiency can present with abnormal uterine bleeding, umbilical bleeding, or severe ovulation bleeding, among other clinical manifestations. However, the authors said factor XIII deficiency can be difficult to diagnose because clotting factor tests and platelet counts are normal in patients with the deficiency.
"The diagnosis can be made by measuring FXIII antigen levels using enzyme-linked immunosorbent assays and/or by measuring its activity with functional methods," the authors said. "Genetic testing of the genes encoding FXIII A or FXIII B is also an option for more specific assessment."
The authors also described several other disorders, including deficiencies of factors V, VIII, X, and XI, and hemophilia.
The investigators said they hope their report serves as a "useful guide" for physicians investigating abnormal uterine bleeding.
"We hope that this review can serve as a roadmap for busy clinicians to familiarize themselves with coagulation factor disorders as a cause of heavy menstrual bleeding and guide them towards more targeted research," they said.
They added, however, that theirs was not a systematic review, and therefore it should not be considered an exhaustive guide to diagnostic decisions.
References
1. Livanou ME, Matsas A, Valsami S, Papadimitriou DT, Kontogiannis A, Christopoulos P. Clotting factor deficiencies as an underlying cause of abnormal uterine bleeding in women of reproductive age: a literature review. Life (Basel). Published online June 5, 2023. Doi:10.3390/life13061321
2. Munro MG, Critchley HOD, Broder MS, Fraser IS; FIGO Working Group on Menstrual Disorders. FIGO classification system (PALM-COEIN) for causes of abnormal uterine bleeding in nongravid women of reproductive age. Int J Gynaecol Obstet. 2011;113(1):3-13. Doi:10.1016/j.Ijgo.2010.11.011
A Spontaneous Cerebral Hemorrhage Case In The Right Temporal Lobe In A Patient With Hemophilia C (XI Factor Deficiency)
@article{heghine_khachatryan_2023_7928161, abstract = {A 28-year-old man was taken to our hospital with sudden onset of severe headache. The computed tomography (CT) scan revealed subcortical hemorrhage of the right temporal lobe. The patient has past medical history of hemophilia C (XI factor deficiency). On neurological examination no deficit was observed (Glasgow Coma Scale (GCS) -15). 10 hours after admission, his GCS and CT scan of the brain were unchanged. After consultation with the hematologist, he received fresh frozen plasma, mannitol, tranexamic acid during the treatment. But 36 hours after admission, the neurological status was worsened (GCS -12)․ A decision was made to immediately perform a temporal craniotomy with evacuation of intracerebral hematoma. Surgery and the postoperative period were uneventful․ The patient was discharged without neurological deficit․ This case highlights the importance of adequate preparation of the patient presurgically as much as possible to minimizes the risk of complications (rebleeding) during the operation and in the postoperative period.}, added-at = {2023-07-14T08:12:00.000+0200}, author = {Khachatryan, Heghine and Harutyunyan, Hayk and Nazinyan, Ruzan and Badalyan, Sevak and Melqumyan, Gurgen and Fanarjyan, Ruben and Hovhannesyan, Yekaterina and Petrosyan, Lilit and Sargsyan, Nelli and Sahakyan, Lusine and Tamamyan, Gevorg}, biburl = {https://www.Bibsonomy.Org/bibtex/2f479c063c78ee647a75b7f1452cf075c/gscarrjournal}, doi = {10.30574/gscarr.2023.14.2.0369}, interhash = {af4a764bcbe8a3ce1c797bfc73dd7aaf}, intrahash = {f479c063c78ee647a75b7f1452cf075c}, issn = {2581-3250}, journal = {GSC Advanced Research and Reviews}, keywords = {Spontaneous cerebral hemorrhage}, month = feb, number = 2, pages = {095–097}, timestamp = {2023-07-14T08:12:00.000+0200}, title = {A spontaneous cerebral hemorrhage case in the right temporal lobe in a patient with hemophilia C (XI factor deficiency)}, url = {https://gsconlinepress.Com/journals/gscarr/content/spontaneous-cerebral-hemorrhage-case-right-temporal-lobe-patient-hemophilia-c-xi-factor}, volume = 14, year = 2023 }
%0 Journal Article %1 heghine_khachatryan_2023_7928161 %A Khachatryan, Heghine %A Harutyunyan, Hayk %A Nazinyan, Ruzan %A Badalyan, Sevak %A Melqumyan, Gurgen %A Fanarjyan, Ruben %A Hovhannesyan, Yekaterina %A Petrosyan, Lilit %A Sargsyan, Nelli %A Sahakyan, Lusine %A Tamamyan, Gevorg %D 2023 %J GSC Advanced Research and Reviews %K Spontaneous cerebral hemorrhage %N 2 %P 095–097 %R 10.30574/gscarr.2023.14.2.0369 %T A spontaneous cerebral hemorrhage case in the right temporal lobe in a patient with hemophilia C (XI factor deficiency) %U https://gsconlinepress.Com/journals/gscarr/content/spontaneous-cerebral-hemorrhage-case-right-temporal-lobe-patient-hemophilia-c-xi-factor %V 14 %X A 28-year-old man was taken to our hospital with sudden onset of severe headache. The computed tomography (CT) scan revealed subcortical hemorrhage of the right temporal lobe. The patient has past medical history of hemophilia C (XI factor deficiency). On neurological examination no deficit was observed (Glasgow Coma Scale (GCS) -15). 10 hours after admission, his GCS and CT scan of the brain were unchanged. After consultation with the hematologist, he received fresh frozen plasma, mannitol, tranexamic acid during the treatment. But 36 hours after admission, the neurological status was worsened (GCS -12)․ A decision was made to immediately perform a temporal craniotomy with evacuation of intracerebral hematoma. Surgery and the postoperative period were uneventful․ The patient was discharged without neurological deficit․ This case highlights the importance of adequate preparation of the patient presurgically as much as possible to minimizes the risk of complications (rebleeding) during the operation and in the postoperative period.
Copy citation to your local clipboard.FDA Approves BioMarin Pharma's Gene Therapy, The First For Hemophilia A
A one-time BioMarin Pharmaceutical treatment that fixes the problem at the root of hemophilia A has finally won FDA approval, making it the first gene therapy authorized for the inherited bleeding disorder.
The Thursday regulatory decision for the gene therapy, Roctavian, covers adults with severe hemophilia A. BioMarin set a $2.9 million wholesale price for the product. But the San Rafael, California-based drugmaker is also giving the hemophilia community and insurance companies assurances that the gene therapy will work. BioMarin is offering a warranty program that will pay back the therapy's price if a patient's disease does not respond to the treatment.
In hemophilia A, patients have an inherited deficiency of factor VIII, a clotting protein. This deficiency makes patients more susceptible to painful bleeding episodes that can happen spontaneously. Standard treatment includes regular infusions of engineered versions of the factor VIII protein that patients lack, which are intended to prevent these episodes. Another option is Hemlibra, a Roche antibody drug that mimics the function of the deficient clotting protein.
Roctavian consists of a functioning version of the gene that provides instructions for making factor VIII. Administered as an intravenous infusion, the gene therapy reaches a patient's cells aboard an engineered virus called adeno-associated virus serotype 5. FDA approval of Roctavian covers patients who don't have pre-existing antibodies to this virus that would render the therapy ineffective. Patients who meet this eligibility requirement are identified with a companion diagnostic that was also approved by the FDA on Thursday. By BioMarin's count, about 6,500 adults in the U.S. Have severe hemophilia A. Of those, the company estimates 2,500 are eligible for Roctavian under the current approval.
The Roctavian regulatory decision comes nearly three years after the FDA rejected the gene therapy, citing data discrepancies between Phase 1/2 testing and the pivotal Phase 3 study. The company continued to follow patients in the study to accrue more data. Approval of Roctavian is based on the results of a Phase 3 study that has more than three years of patient data. Of 134 patients who received the gene therapy, 112 had annualized bleeding rate data collected for six months prior to receiving Roctavian. During this period, they received factor VIII therapies. The remaining 22 patients had their baseline data collected retrospectively.
The trial results showed that following dosing with Roctavian, patients' annualized bleeding rate dropped to 2.6 episodes per year, a 52% decline from baseline. In addition, patients also reported a reduction in the rate of spontaneous bleeds and joint bleeds compared to baseline. The most common adverse reactions included nausea, fatigue, and headache. But laboratory testing also showed higher levels of liver enzymes, which can be a sign of toxic effects on the liver. This problem can be addressed with corticosteroids. The drug's label tells physicians to monitor a patient's liver enzymes weekly for at least 26 weeks after dosing, and to monitor and manage for the any adverse effects associated with corticosteroid treatment.
Earlier this week, during the annual meeting of the International Society on Thrombosis and Haemostasis, BioMarin presented three-year data showing that participants in the Phase 3 study had an 82.9% reduction in treated bleeds overall compared with baseline. The study also found the gene therapy led to a 96.8% reduction in factor VIII usage overall compared with baseline.
"Spontaneous bleeds and joint bleeds are medically important events that define severe hemophilia A as a bad condition, and the fact that those are dramatically affected by Roctavian is Roctavian's superpower," Hank Fuchs, BioMarin's president of worldwide research & development said, speaking during a conference call Thursday.
The European Commission granted Roctavian conditional marketing authorization last August, based on two-year clinical trial data. While Roctavian is the first gene therapy approved for hemophilia A, it is not the first hemophilia gene therapy. Last November, the FDA approved Hemgenix, a CSL Behring gene therapy for the rare hemophilia B. Hemgenix carries a $3.5 million price, but CSL Behring has pledged to offer value-based agreements to commercial payers. These agreements tie the reimbursement of a therapy to predetermined outcomes that show the treatment worked.
Under the warranty for Roctavian, BioMarin will reimburse up to 100% of the therapy's wholesale acquisition cost for those who don't respond to the therapy. BioMarin Chief Commercial Officer Jeff Ajer said this four-year warranty will be offered to all U.S. Insurers, both public and private. If a treated patient loses response to the therapy within the four-year period, Ajer said BioMarin will reimburse payers on a prorated basis. But he added that continued benefit beyond four years represents savings to the healthcare system.
Ajer said Roctavian will become commercially available in the U.S. In about two months. But prior to receiving the therapy, patients will need to go through several steps that include determining if their insurance will cover the therapy, taking the test to determine eligibility for the treatment, and testing to establish a baseline for liver enzyme levels. Patients should expect that the various steps will take anywhere from two to five months, Ajer said.
Public domain image by Flickr user SciTechTrend


Comments
Post a Comment