Hematological Disorders

What To Know About Hemophilia B
Hemophilia B, also known as Christmas disease, is a bleeding disorder. Its characteristic feature is a deficiency of clotting factor IX. This is a protein present in the blood that helps with coagulation, or clotting.
Hemophilia describes a group of bleeding disorders where blood is unable to clot properly. This occurs due to a deficiency in certain proteins known as clotting factors. Many different clotting factors play a different role in the coagulation cascade.
The coagulation cascade refers to a series of steps that occur in response to bleeding. The process involves clotting factors, and the series of reactions ultimately leads to the formation of a blood clot. A deficiency in a clotting factor impairs this process and can result in spontaneous bleeding or severe bleeding following an injury.
Hemophilia B primarily occurs due to a genetic variation in the F9 gene, which is present on the X chromosome. This is why medical professionals may describe hemophilia B as an X-linked condition.
The F9 gene is responsible for producing factor IX. As such, this gene alteration results in a deficiency, or impaired function, of clotting factor IX.
Clotting factor IX is one of the many clotting factors that play a crucial role in the blood clotting cascade. Without sufficient levels of factor IX, a person will experience difficulties forming blood clots and is susceptible to prolonged bleeding.
In most cases, people inherit the gene alteration for hemophilia B from a parent. However, in extremely rare cases, a person may develop acquired hemophilia B. This type of hemophilia is not present at birth — instead, an individual develops the condition later in life.
Acquired hemophilia B occurs when the body mistakenly produces antibodies against factor IX. The factor IX antibodies then destroy any circulating factor IX in the blood, resulting in the bleeding symptoms of hemophilia.
Hemophilia is a group of conditions that occur due to problems with clotting factors. There are multiple clotting factors and different types of hemophilia, which correspond to deficiencies of different clotting factors.
Hemophilia A is the most common type of hemophilia. Its characteristic feature is a deficiency of factor VIII due to variations in the F8 gene. Hemophilia C is also distinct from hemophilia B, as it instead occurs due to alterations in the F11 gene and affects clotting factor XI. There are also less common conditions that affect other blood clotting factors.
Hemophilia conditions typically share similar symptoms and treatment approaches but result from variations in different genes that affect different clotting factors.
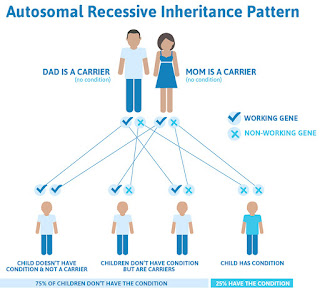
Similar to other types of hemophilia, hemophilia B is an inherited bleeding disorder primarily affecting males. Evidence notes that the prevalence of hemophilia B is roughly 1 in 40,000 live males. This accounts for about 15% of cases of hemophilia.
Hemophilia B is typically a congenital condition, meaning people are born with it. In most cases, a person inherits the condition from a parent.
The F9 gene is present on the X chromosome. A male will usually inherit one X chromosome from a female parent, while females typically inherit an X chromosome from both parents. As such, males usually only require one gene alteration to develop the condition, which is why it is more common in males.
While much rarer, hemophilia can also affect females. They will typically require either two X chromosome alterations, or one with a variation and the other X chromosome is missing or nonfunctioning.
Evidence also notes that roughly 10–25% of female carriers for hemophilia B may also experience symptoms of the condition. Hemophilia B also affects all races and ethnic groups equally.
Some people may also refer to hemophilia B as the royal disease due to being prevalent in the royal families of England, Spain, Germany, and Russia.
Read on to learn more about how people inherit hemophilia.
The characteristic symptom of hemophilia is issues with blood clotting. This makes people with the condition more susceptible to prolonged periods of bleeding.
The severity of hemophilia B can range from mild to severe. The severity depends on the level of factor IX present in the blood. Symptoms of hemophilia B can include:
Severe hemophilia BThis describes a person with less than 1% clotting activity. They may experience:
This typically refers to individuals with 1–5% clotting factor activity. Symptoms may include:
Mild hemophilia B usually describes factor activity between 5% and 40%. People may experience:
Most people with severe hemophilia B will likely receive a diagnosis at birth. Individuals with moderate or mild hemophilia B may receive a diagnosis during adolescence or childhood.
The diagnosis of hemophilia B usually involves blood tests. These tests will measure the levels and activity of clotting factors. As such, the tests can also help determine the severity of hemophilia B. These may include:
A doctor may recommend genetic testing and counseling for individuals with a family history of hemophilia. These tests can help identify how likely a person is to potentially pass hemophilia to their children.
The Centers for Disease Control and Prevention (CDC) notes that hemophilia can lead to the following complications:
A person living with hemophilia may also develop inhibitors. This term refers to antibodies the immune system may produce, which attack replacement clotting factors a person receives to treat hemophilia. As such, inhibitors may make it more difficult to treat bleeding episodes.
Similar to treating other types of hemophilia, the primary treatment option for hemophilia B involves replacing factor IX. A person will usually receive replacement therapy through regular intravenous infusions through a vein in the arm or a port in the chest.
Before surgery or dental procedures, a doctor may also recommend aminocaproic acid. It is an antifibrinolytic, which means it prevents the breakdown of blood clots. As such, it may help to preserve a blood clot and keep it from breaking down prematurely.
Etranacogene dezaparvovec (Hemgenix) is a gene therapy that received approval from the Food and Drug Administration (FDA) in 2022 for treating hemophilia B in adults. The treatment uses a modified virus that contains copies of the F9 gene.
After injecting Hemgenix into a person's vein, the virus travels to the liver, which allows the liver cells to produce factor IX and limit bleeding episodes.
Hemophilia B is a bleeding disorder. It typically occurs due to a variation in the F9 gene, which a person inherits from their parents.
Alterations in the F9 gene affect the production of clotting factor IX. This protein plays an important role in the formation of blood clots. Without sufficient levels of factor IX, a person is susceptible to long bleeding episodes.
Hemophilia B can vary in severity, depending on the levels present in the blood. Treatment for hemophilia B usually involves replacing the deficient factor IX to help with blood clotting and prevent excessive bleeding.
Evaluating WHO's Hemophilia Treatment Choices: Patient Safety, Access Challenges
A commentary questions the characterization of cryoprecipitate, which is not pathogen-reduced, as an alternative modality for treating hemophilia, despite its substantial risk of transmitting blood-borne pathogens to patients.
Red Blood CellsImage Credit: Design Cells - stock.Adobe.Com
Recent decisions made by the World Health Organization (WHO) regarding the inclusion and categorization of treatments for hemophilia in its Essential Medicines List (EML) have sparked concerns among patients and health care providers.
One of the primary concerns revolves around the characterization of cryoprecipitate (cryo), which is not pathogen-reduced (PR), as an alternative modality for treating hemophilia, despite its substantial risk of transmitting blood-borne pathogens to patients.
The decision to include cryo in the EML raises questions about patient safety and the potential transmission of infections in countries where blood safety standards might not be as stringent, according to a commentary article published in Haemophilia by Albert Farrugia, PhD, University of Western Australia.
PR-cryo: Affordable, Precise Treatment
Cryo was the first therapeutic product available for treating bleeding disorders and has long been utilized for conditions like hemophilia A, von Willebrand's disease, and fibrinogen deficiency. However, in developed countries, the unmodified version is considered outdated, with specific concentrates of the deficient proteins being the preferred mode of treatment.
The inclusion of cryoprecipitate [pathogen-reduced] (PR-cryo) in the EML is especially relevant for resource-poor conditions. PR-cryo, specified for Factor VIII (FVIII), von Willebrand Factor, and fibrinogen, offers a more affordable and accurate dosing option.
Pooled and aliquoted from individual cryoprecipitates, PR-cryo ensures the potency of the relevant factors, allowing for precise and effective treatment. This method is pivotal in areas where resources are limited, ensuring patients receive the necessary care without the burden of excessive costs.
The WHO Global Status Report on Blood Safety and Availability highlights a significant disparity in blood safety between low- to middle-income countries (LMIC) and high-income countries (HIC). It's reported that LMICs face a much higher prevalence of blood-borne viruses, posing a considerable risk to patients relying on blood products for treatment.
The commentary discusses how the availability of several commercial versions of PR-cryo might impact the cost-effectiveness of the therapeutic modality. Coagulation factor concentrates, especially for hemophilia A, have become more affordable in recent years, benefiting developing countries.
However, Farrugia referenced a recent report that suggests domestically sourced PR-cryo might offer a cheaper alternative demonstrating the importance of ongoing research and international cooperation to ensure the most cost-effective solutions reach those in need.
Categorization of Hemophilia Treatments
The decision to categorize FVIII and factor IX (FIX) concentrates as complementary rather than core medicines has raised concerns within the medical community. While these concentrates are recognized as the preferred route of replacement therapy for hemophilia A and B, their listing status might be influenced by cost perceptions, leading to reservations among health care providers.
Patients with hemophilia B, in particular, could experience disadvantages if a core-listed medicine is not specified, calling for careful reconsideration of these classifications to ensure equitable access to essential treatments.
Reconsidering Alternative Treatments
Another area of concern was noted from the inclusion of Prothrombin Complex Concentrate (PCC) as an alternative treatment to FIX concentrates for hemophilia B. Despite its historical use, evidence-based practice has shown that PCC does not align with the advancements made in hemophilia care.
This decision to list PCC as an alternative treatment, Farrugia wrote, contradicts established medical practices and warrants urgent reevaluation to ensure patients receive the best and safest treatments available.
The commentary acknowledged the WHO's recognition of the value of blood products in hemophilia treatment but stated there is a pressing need for a comprehensive review of the EML in the context of evolving medical advancements. Incorporating safer, more effective treatments while addressing the economic challenges faced by low-income countries should be at the forefront of decision-making.
Reference
Farrugia, A. The World Health Organisation's list of essential medicines and haemophilia treatment products. Haemophilia. 2023; 1-3. Https://doi.Org/10.1111/hae.14879
What Are Dental Veneers? Benefits, Cost And More
Having dental veneers put on is a relatively simple procedure performed in your dentist's office. However, you may need to make a few appointments to complete the entire process, says Samantha Rawdin, D.M.D., a prosthodontist based in New York. Typically, you'll have a consultation, then a follow-up or two for fitting and applying. Below is a breakdown of the steps involved:
Initial Consultation"The first step for diagnosis involves an intimate consultation between the dentist and patient," says Dr. Rubinov. "I always ask my patients to explain to me in detail the insecurity they have with their smile. "This helps me come up with a plan that produces the result that they are trying to achieve."
The initial appointment will likely involve a physical exam to assess your overall oral hygiene, spot any issues (like excessive tooth decay) before veneers are applied and discuss your goals, says Dr. Rawdin. "You should also expect to have X-rays and photos taken, and possibly even impressions of your teeth," she says.
Preparing Your TeethOnce both you and your dentist have decided you're a good candidate for veneers, your dentist will prepare your teeth. This can involve fixing cavities, reshaping the surface of your teeth and "roughing" the enamel to help the cement adhere better, says Dr. Rubinov.
Fitting and Bonding the VeneersOnce your custom veneers come back from the lab, which Dr. Rawdin says can take several weeks, you'll go in for a fitting appointment. Here, your dentist will test the size, color and overall appearance. If everything looks and feels right, your veneers will be bonded right away. If not, your veneers may need to be sent back to the lab for adjustments.
AftercareGetting dental veneers isn't overly painful and there isn't much of a recovery period. "In the hours following your procedure, your gums might be sore from the area that we administered local anesthesia," says Dr. Rubinov, "but the next day you can expect to love your new smile." If you do experience discomfort afterward, over-the-counter medication can help, he adds.
Your Smile, For Life
You own your smile, and you should own it forever. With Byte, watch your smile transform with digital access to your doctor-directed treatment plan.
$80 Off With Code FORBESBBP

Comments
Post a Comment