MLB Weekly Digest September 3rd Edition - NGSC Sports
Everything To Know About Hemophilia B
Hemophilia B is also known as Christmas disease. It's a rare genetic blood clotting disorder that can be fatal without treatment.
A person is born with hemophilia B, but it may not be diagnosed until later in life. It's estimated that two-thirds of cases are inherited, according to the National Hemophilia Foundation.
The other cases are caused by spontaneous gene mutations that occur for unknown reasons during fetal development. The disease occurs almost exclusively in people who are male.
Read on to learn what causes hemophilia B, its symptoms, and how it's treated.
Hemophilia B is a type of hemophilia, with the other types being hemophilia A and hemophilia C.
It is also known as factor IX hemophilia and is a rare genetic disorder in which your blood does not clot properly.
The condition is also sometimes called Christmas disease after Stephen Christmas, who was the first person diagnosed with the condition in 1952.
If you have hemophilia B, your body produces little or no factor IX, a protein that helps the blood clot. This leads to prolonged or spontaneous bleeding. The less factor IX your body produces, the worse your symptoms are. Without treatment, hemophilia B can be fatal.
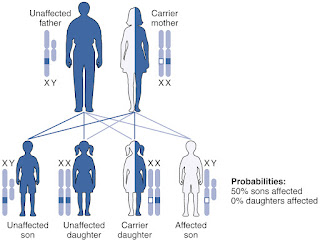
The gene responsible for hemophilia B is carried on the X chromosome.
Male children inherit one X and one Y chromosome, while female children inherit two X chromosomes. If a male child inherits a mutated gene on his X chromosome from his female parent, he will develop hemophilia B.
On the other hand, female children with one mutated gene are carriers and can pass the gene to their children. Male parents with the mutated gene will always pass it on to their female children but not their male children.
In rare cases, both parents may have the mutated gene, but this is uncommon.
With each pregnancy, female carriers have a:
Severe cases of hemophilia B are usually diagnosed in babies younger than one year old. Mild cases may not be diagnosed until a child reaches their toddler years or sometimes even adulthood.
In all cases, diagnosis usually happens after abnormal bleeding from an injury or surgery.
Events that may lead your doctor to suspect hemophilia B include:
Severe cases of hemophilia B may cause unexplained bleeding in the skull after childbirth and spontaneous bleeding.
If you or your child shows symptoms of hemophilia B, your doctor may order blood screenings and tests to confirm the diagnosis. These may include:
If you're female with a family history of hemophilia B, you can have genetic testing to see if you carry the faulty gene. Genetic testing is a very accurate way to detect the responsible gene.
There's no cure for hemophilia B, but there are treatments for the condition. Regular treatment is essential for managing the symptoms of hemophilia B.
Factor IX injectionsHemophilia B can be treated with factor IX injections to prevent or stop bleeding. The factor IX can be derived from donated human blood or made in a laboratory.
Artificial factor IX is called recombinant factor IX and is recommended over blood-derived factor because it's safer.
Blood-derived factor IX is rarely used in the United States.
Preventive treatmentIf you have a severe form of hemophilia B, you may need preventive blood transfusions to avoid or reduce prolonged and heavy bleeding, which is known as prophylaxis. These are especially important in children. If you receive blood-derived factors or blood transfusions, you should be vaccinated for hepatitis B.
People with severe hemophilia B are at a slight risk of:
Treatment for hemophilia B can sometimes lead to abnormal blood clots. To prevent complications, get regular checkups and blood tests, and avoid aspirin and other platelet-acting medications.
A small percentage of people with hemophilia B will develop antibodies (known as inhibitors) against factor IX replacement therapy.
These antibodies destroy the replacement factor in factor IX injections, causing this treatment to be ineffective. Your doctor can test for your risk of developing inhibitors and prescribe alternative treatments if necessary.
When you first receive a diagnosis of hemophilia B, you may feel confused and overwhelmed.
Your healthcare professional is there to answer any questions you may have that would help you cope as you learn to live with the condition and receive treatment. Some questions you can ask include:
In people with mild-to-moderate hemophilia, regardless of type, the death rate is 19% higher than in healthy people.
That said, life expectancy is continuing to improve. With treatment, most people with hemophilia B are likely to lead normal lives.
It's important to make sure you avoid situations in which excess bleeding could occur. You also can receive blood clotting therapy before any surgery or after any injury.
Learn more about the life expectancy of people with hemophilia.
How serious is hemophilia B?Without management, hemophilia can be life threatening, especially due to accidents or injuries that could lead to excessive bleeding. Talk with your doctor about ways you can prevent bleeding, tips on how to manage your condition if an injury occurs, and other supportive resources.
Can hemophilia B be cured?There is no cure for hemophilia B, but the condition can be managed with treatment.
Why is hemophilia B called royal disease?In addition to Christmas disease, hemophilia B is sometimes called the royal disease because a number of European royals in history are known to have either carried the gene for the disease or had the condition. For example, Queen Victoria of England was believed to be a carrier. Alexei Nikolaevich, the son of Emperor Nicholas II and Empress Alexandra Feodorovna of Russia, had the disease.
Hemophilia B, also known as factor IX hemophilia, is one of three types of hemophilia. It's a rare genetic disorder that causes problems with blood clotting. It was first identified in 1952 by Stephen Christmas, who was diagnosed with it.
The severity of symptoms varies depending on the amount of factor IX produced by the body, but without proper treatment, hemophilia B can be life threatening.
Read this article in Spanish.
Novel Monoclonal Antibody Reduces Bleeds In Hemophilia A And B
SAN DIEGO -- The investigational monoclonal antibody marstacimab reduced the rate of treated bleeds in patients with hemophilia A or B without inhibitors to factor VIII or factor IX, the phase III BASIS trial showed.
The annualized bleeding rate (ABR) for treated bleeds was reduced by 91.6% with marstacimab versus on-demand therapy with factor VIII or factor IX concentrates, and by 35.2% compared with routine prophylactic care, reported Davide Matino, MD, MSc, of McMaster University in Hamilton, Ontario.
"Marstacimab was effective and safe for reducing bleeds in participants with severe hemophilia A or moderate to severe hemophilia B without inhibitors at 12 months in the phase III study, and up to an additional 16 months in the long-term extension study," Matino said during a session at the American Society of Hematology annual meeting.
Pfizer, the drug's developer, announced Monday that based on efficacy and safety data from the BASIS trial, the FDA has accepted the biologics license application for marstacimab, with a decision date set for the fourth quarter of 2024.
According to the company, if approved, marstacimab is expected to be the first once-weekly subcutaneous treatment for people living with hemophilia B, and the first treatment administered as a flat dose for people living with hemophilia A or B.
Marstacimab is targeted to the tissue factor pathway inhibitor protein to improve hemostasis via the extrinsic pathway of blood coagulation. Previous phase I/II studies demonstrated the efficacy and safety of long-term administration of marstacimab up to 450 mg weekly for reducing bleeding episodes in adults with severe hemophilia A or hemophilia B, with or without inhibitors, compared with on-demand therapy.
In the BASIS trial, 128 participants (108 adults and 20 adolescents) with hemophilia A or B entered a 6-month observational phase following screening and were categorized according to factor replacement treatment -- either an on-demand (37 patients) or routine prophylaxis (91 patients) intravenous regimen of factor VIII or factor IX administered as part of usual care.
Of these patients, 116 completed the observational period (33 on-demand and 83 routine prophylaxis) and crossed over to a 12-month active treatment period, receiving a single subcutaneous loading dose of 300 mg followed by once-weekly 150-mg marstacimab.
Among the patients who received treatment on-demand, the ABR for treated bleeds fell from a mean of 38 during the observational period to 3.2 after treatment with marstacimab, representing a 91.6% reduction in mean ABR during the active treatment period (P<0.0001). Ten of 33 participants had no treated bleeds with marstacimab.
Following the 12-month active treatment period, patients could continue to receive marstacimab in a long-term extension study, which showed that efficacy was maintained (ABR 0f 3.9) for up to an additional 16 months.
Among the patients who received routine prophylaxis as usual care, the ABR fell from a mean of 7.9 during the observational period to 5.1 after treatment with marstacimab, translating into a 35.2% reduction in mean ABR during the active treatment period, demonstrating non-inferiority and superiority (P=0.0376). About a third of patients did not have any treated bleeds with marstacimab. Efficacy was also maintained during the long-term extension in this group, with an ABR of 2.3.
Matino reported that the efficacy of marstacimab was generally consistent across all subgroups, including patients with hemophilia A and B, as well as in adolescents and adults.
Marstacimab also showed superiority compared with previous on-demand therapy for joint bleeds, spontaneous bleeds, target joint bleeds, and total bleeds (all P<0.0001), while marstacimab was shown to be non-inferior to routine prophylaxis with factor concentrates across these bleed categories.
There were also non-significant improvements in health-related quality-of-life parameters with marstacimab compared with on-demand therapy, and non-inferiority versus routine prophylaxis therapy.
Most adverse events were mild to moderate in severity, Matino reported, and no embolic or thrombotic events were observed with marstacimab in either the main or extension phase of the study.
Mike Bassett is a staff writer focusing on oncology and hematology. He is based in Massachusetts.
Disclosures
The study was funded by Pfizer.
Matino reported relationships with Bayer, Pfizer, Novo Nordisk, Sanofi, Sobi, Spark, Octapharma, and Roche.
Primary Source
American Society of Hematology
Source Reference: Matino D, et al "Efficacy and safety of the anti-tissue factor pathway inhibitor marstacimab in participants with severe hemophilia without inhibitors: results from the phase 3 BASIS trial" ASH 2023; Abstract 285.
Please enable JavaScript to view the comments
Gene Therapy: Redefining Hemophilia Treatment
PHILADELPHIA, Pa. (Ivanhoe Newswire) – Hemophilia – once a death sentence, is now on the verge of having a cure. An inherited disorder, mostly affecting boys, happens when blood doesn't clot. Hemophilia can cause spontaneous and severe bleeding following an injury. Twenty years ago, there were only treatments to stop the bleeding. Now, gene therapies hold even more promise of one day curing this disease.
The Ward brothers may have different interests, but Jadon and Roan have more in common than you can see.
"Hemophilia is when you have a lot of bleeding," Roan says.
"My blood doesn't clot like the average human," Jadon explains.
Both were born with hemophilia.
Their mother, Melody Ward, says, "Definitely, a bleed is the biggest threat to them."
"When I started 50 years ago, most of the patients with hemophilia had some kind of deformity or disability. You know, some were in wheelchairs, some wore braces, some were in crutches," explains Regina Butler, RN, a hematology clinical manager at the Children's Hospital of Philadelphia.
Butler has treated five generations of Melody's family, including her father, uncles and now her sons, who take shots every few weeks to prevent bleeding.
"The treatment has evolved so rapidly. We kept getting better and better products," Butler expresses.
Now, a new gene therapy has been approved by the FDA for Hemophilia B. Through a one-time IV infusion, Hemgenix instructs the body to create functional factor nine genes that will help the blood to clot.
Butler says, "It's remarkable to me. I feel like I've come full circle in my career with hemophilia."
Hemgenix can only be administered once, but its impact can last for years, making the injections the Ward boys take, obsolete.
"I didn't think that there would be a time where I would say there was a cure for hemophilia," Melody exclaims.
The treatment is currently approved for patients 18 years and older, but doctors hope in the future to be able to treat children as young as 12. The most common adverse reactions associated with Hemgenix include liver enzyme elevations, headaches, and flu-like symptoms.
Copyright 2023 WALB. All rights reserved.



Comments
Post a Comment