Childhood Cancer Epidemiology: Overview, Tools of Study, Cancer Incidence
Sickle Cell Disease Might Have Contributed To Death Of Student In Racially Charged Chase: Experts
Justin Johnson, 16, died Friday April 26, 2024.Orville Johnson
The sudden death of 16-year-old Central Dauphin student Justin Johnson after being chased by other students from his school could have been connected to the boy's sickle cell disease, according to experts.
Sickle cell disease condition is a blood disorder that leaves some red blood cells in a rigid, C-shape, like the farm tool called a sickle, rather than their usual, flexible, round shape.
If you purchase a product or register for an account through a link on our site, we may receive compensation. By using this site, you consent to our User Agreement and agree that your clicks, interactions, and personal information may be collected, recorded, and/or stored by us and social media and other third-party partners in accordance with our Privacy Policy.
Innovation Sought For Fertility Tests In Young Males With Sickle Cell
Having male adolescents with sickle cell disease (SCD) undergo fertility testing by semen analysis may not be feasible if it involves travel to a reproductive diagnostic center, according to a recent study.
Despite having access to reproductive health education and compensation to cover transportation costs, only 15% of adolescents participating in the study adhered to fertility testing. Barriers to testing included discomfort and a lack of time or transportation.
"These findings further underscore the need for innovative strategies to examine semen parameters and fertility in male youth with SCD to ensure that they are able to achieve their health and reproductive goals," the researchers wrote.
The study, "Experiences and outcomes of fertility testing in male adolescents with sickle cell disease," was published in the journal Pediatric Blood & Cancer.
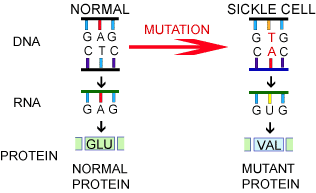
SCD is caused by mutations in the beta-globin (HBB) gene, which contains instructions to make a component of hemoglobin — the protein in red blood cells that's responsible for transporting oxygen throughout the body.
These mutations ultimately result in the production of a defective form of hemoglobin that causes red blood cells to take on the shape of a sickle. Misshapen red blood cells cannot pass through small blood vessels as easily, slowing or obstructing blood flow and impairing oxygen delivery to body tissues.
Recent studies have shown that adult males with sickle cell receiving or not receiving hydroxyurea, an approved treatment for sickle cell, have abnormal semen parameters, raising the concern that the disease and/or its treatments might affect fertility.
"Yet, studies that include adolescents are lacking," the researchers wrote.
The study and its findingsIn this study, researchers in the U.S. Sought to determine the feasibility of obtaining semen samples from adolescents with sickle cell at a reproductive diagnostic center as well as the barriers associated with testing. It also sought to examine the semen parameters of adolescents who completed the procedure.
For that, they recruited adolescents with sickle cell, ages 14 to 22, and their caregivers to participate in the study. They were asked to complete a web-based reproductive health program called Fertility edUcation To Understand Reproductive hEalth in SCD (FUTURES). The program described the potential impact of sickle cell and/or its treatments on fertility, as well as the purpose and process of fertility testing.
A total of 33 adolescents completed FUTURES. After that, they were offered the option to have a semen analysis at a reproductive diagnostic center, with costs of testing and transportation covered. Fertility testing was considered to be feasible if at least 25% of the adolescents who completed FUTURES opted to be tested.
Overall, five adolescents (15%) submitted a sample for semen analysis, a proportion that was below the minimum to consider fertility testing as feasible.
Reasons pointed out by adolescents and their caregivers to complete semen testing included interest in learning about their reproductive health, assistance in coordinating appointments, and the absence of costs. The reasons for not testing included discomfort, lack of time or transportation, and forgetting the appointment.
Two of the adolescents who underwent fertility testing had the HbSC disease subtype, and three had the HbSS subtype. Two of those with HbSS were being treated with hydroxyurea.
HbSS, usually the most severe form of sickle cell disease, occurs when patients have both mutated HBB gene copies producing a faulty version of hemoglobin called hemoglobin S. The HbSC subtype occurs when one gene copy produces hemoglobin S while the other encodes hemoglobin C, another defective form of the protein.
Each analysis provided information on several semen parameters, including volume, viscosity, sperm cell levels, motility (movement), and morphology (form and structure).
Sperm cells were identified in the three adolescents who had never been treated with hydroxyurea. In two of them, sperm cell levels were within the normal range; in the third, they were considered low. On the contrary, in the two adolescents then receiving hydroxyurea, very few or no sperm cells were found.
According to researchers and considering the small sample in this study, "more research is needed to elucidate these effects (e.G., what effect SCD and/or hydroxyurea has, whether or not these effects may be reversible) and examine actual fertility outcomes."
"Our qualitative findings suggest that developmentally and culturally appropriate fertility education and increased advocacy for affordable reproductive healthcare for this population may facilitate adolescents with SCD to pursue fertility testing, whereas innovative methods to collect semen samples that limit patient discomfort and time constraints (e.G., mail-in testing where semen samples are collected at home) are needed to reduce barriers," they wrote.
CASGEVY Gene Therapy Eliminates Vaso-occlusive Crises In Sickle Cell Patients
In a landmark study, an international consortium led by researchers at Children's Hospital of Philadelphia (CHOP) published the final results of a key clinical trial of the gene therapy CASGEVY (exagamglogene autotemcel) for the treatment of sickle cell disease in patients 12 years and older with recurrent vaso-occlusive crises (VOCs). The study found that 96.7% of patients in the study did not have any vaso-occlusive crises (VOCs) – a blockage that results in lack of oxygen and painful episodes – for at least one year, and 100% were able to remain hospitalization-free for the same length of time.
The findings, published today in the New England Journal of Medicine, provide the complete details of the critical clinical trial that led to the FDA approval of CASGEVY™ for the treatment of sickle cell disease in December 2023.
Sickle cell disease is a lifelong condition that causes intense pain due to deformed blood cells that can cause blockages in blood vessels. This can also lead to strokes, organ damage, and shortened lives.
Researchers have been studying the use of gene therapy and CRISPR technology to edit portions of DNA in people with inherited or genetic disorders, like sickle cell disease. In the case of sickle cell disease, the CASGEVY process edits DNA within the patient's own cells and enables the patient to produce a different form of hemoglobin in their red blood cells. Clinical trials at CHOP and other sites have shown that successful gene editing can prevent cells from developing the distinctive crescent shape apparent in sickle cell disease and have eliminated pain episodes in almost all patients. CASGEVY was the first FDA-approved therapy developed with CRISPR technology.
"In this clinical trial, sickle cell patients who were having significant issues with their disease began to see their problems resolve within months and improve their quality of life significantly," said senior study author Stephan A. Grupp, MD, PhD, Section Chief of the Cellular Therapy and Transplant Section, Inaugural Director of the Susan S. And Stephen P. Kelly Center for Cancer Immunotherapy, and Medical Director of the Cell and Gene Therapy Laboratory at CHOP. Grupp was also one of the principal investigators in the clinical trials that led to the approval of CASGEVY and the leader of the study's steering committee.
The researchers conducted the CLIMB SCD-121 trial, a phase 3, single-arm, open-label study of exa-cel in patients between 12 and 35 years old with sickle cell disease and at least two severe VOCs in each of the two years before screening. The key primary endpoint of the study was a proportion of patients without severe VOCs for at least 12 consecutive months, with a secondary endpoint of patients who were free from inpatient hospitalization for severe VOCs for at least 12 consecutive months.
A total of 44 patients received exa-cel with a median follow up of 19.3 months. In a total of 30 patients with sufficient follow-up data to be evaluated, 29 (96.7%) were free of VOCs for at least 12 consecutive months. This information is an update for the US Prescribing Information for CASGEVY, which includes an evaluation of 31 patients resulting in a response rate of 93.5%. The safety of treatment was comparable to treatment with hematopoietic and progenitor stem cells, and no malignancies were reported as a result of treatment.
This study was supported by Vertex Pharmaceuticals and CRISPR Therapeutics.
Source:
Journal reference:
Frangoul, H., et al. (2024) Exagamglogene Autotemcel for Severe Sickle Cell Disease. New England Journal of Medicine. Doi.Org/10.1056/NEJMoa2309676.



Comments
Post a Comment