Hypogonadism Natural Treatment: Tips for Men and Women
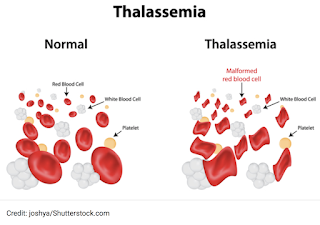
Complications Of Beta Thalassemia
The blood disorder beta thalassemia can bring complications that include things like bone damage, heart problems, and slow growth in children. Treatment can help you or your child avoid many of these problems.
Beta thalassemia lowers the amount of a protein in your body called hemoglobin. Hemoglobin helps red blood cells carry oxygen to your organs and tissues. If you or your child doesn't have enough hemoglobin, you can get anemia, which makes you tired and short of breath.
Low oxygen and too much iron cause most beta thalassemia complications. Iron is a mineral your body uses to carry oxygen and keep your muscles healthy. In beta thalassemia, your intestines absorb more iron than normal. The blood transfusions you get to treat the disease also contain iron. All of that extra iron builds up in your heart, liver, and the glands that make hormones and damages these organs.
The complications you or your child get depend in part on the type of beta thalassemia. "Beta thalassemia minor" is mild and usually doesn't cause problems. Anemia from "beta thalassemia intermedia" causes slowed growth in children, weak bones, and an enlarged spleen. "Beta thalassemia major" is the most serious type, and it can cause many complications, including slow growth in children, an enlarged spleen, heart and liver problems, and bone damage.
If you're the parent of a child who has complications from beta thalassemia, talk to your friends and family to get the emotional backing you may need as you help your child manage their symptoms. If you find yourself getting anxious or stressed out, talk to your doctor. They can put you in touch with social workers or mental health professionals who can help.
Your child's body needs lots of energy to grow. Cells need oxygen to create that energy.
Without enough oxygen, a child will grow more slowly. Puberty may also start late in kids with beta thalassemia.
Your spleen is an organ that makes new infection-fighting white blood cells and filters out the old ones that your body doesn't need anymore. Beta thalassemia makes your spleen make more new blood cells and break down more old blood cells than usual, so it has to work harder.
Just like a muscle grows when you use it more, overuse makes your spleen get bigger. If your spleen gets too large, you may need an operation called a splenectomy to remove it.
Your spleen is part of your body's defense system against germs. It makes the white blood cells that protect you from infections.
An enlarged spleen doesn't work as well as it should, which could make you more likely to get sick. And if you have surgery to remove your spleen, you or your child will be more likely to catch infections like the flu and pneumonia.
Getting all of your recommended vaccines and taking antibiotics will help to protect you from some of these illnesses. Let your doctor know if your child with beta thalassemia runs a fever. This could be a sign of a serious infection.
In severe beta thalassemia, both anemia and iron overload can damage the heart and cause problems like:
Heart problems can get worse without causing any symptoms. You or your child should get tests like echocardiograms, a chest X-ray, and a stress test each year to watch for any problems. Medicine to remove extra iron in your body, called chelation therapy, can help prevent heart problems from too much iron.
Your liver helps keep the right balance of iron in your body. Extra iron due to beta thalassemia or blood transfusions can build up and damage the liver.
Although it's rare, it is possible to get hepatitis B from a blood transfusion, which can also damage the liver.
Eventually your liver can become so scarred that it doesn't work right, a condition called cirrhosis.
Your body makes new blood cells in bone marrow, the spongy area inside your bones. When you have anemia, your bone marrow has to work overtime to make enough red blood cells to meet your body's needs.
As the bone marrow works harder, it stretches. Your bones become thinner, wider, and weaker than usual. They can break easily.
Extra bone growth may also cause your forehead, cheekbones, or jaw to stick out more than usual.
Your doctor will monitor you or your child for complications and treat any problems. One way to avoid complications is to follow the treatment plan your doctor prescribed.
If you have severe beta thalassemia, blood transfusions can help you avoid some of these problems.
Chelation therapy helps prevent extra iron from damaging your organs. You get this treatment as a pill or shot. The medicine binds to iron in your body and removes it through your urine or a bowel movement.
Different Types Of Thalassemia, Their Characteristics, And Treatment Options Explained
© Provided by The Indian Express thalassemia Launched by the Thalassemia International Federation, the day also aims to promote and strengthen the morale of the survivors who have battled the fatal disease for years. (Source: Freepik)
Every year, World Thalassemia Day is observed on May 8 to raise awareness about thalassemia, a chronic blood disorder, among the public and policymakers. Launched by the Thalassemia International Federation, the day also aims to promote and strengthen the morale of those who have battled the fatal disease for years.
What is thalassemia?
Thalassemia is an inherited genetic hemoglobinopathy, a group of disorders that lead to defective production of haemoglobin synthesis in the body. This results in low production of red blood cells and a lack of oxygenated blood supply to the body parts, explained Dr Preetam Jain, Medical Oncologist, Bhatia Hospital, Mumbai.
Adding that thalassemia is an inherited blood disorder, meaning at least one of the parents must be a carrier of the same, the expert said that it is caused by "either a genetic mutation or a deletion of certain key gene fragments." "There is diminished or absent production of globin chains. This results in imbalanced globin chain production, which leads to tetramers and chronic hemolysis (premature destruction and shortened red blood cell life span)," he told indianexpress.Com.
While some of the common symptoms include anemia, fatigue, enlarged liver and spleen, growth impairment, skeletal deformities, leg ulcers and infections, hemolytic facies, frequent transfusions, and iron overload, experts pointed out that these can vary. "Additionally, anybody can develop this genetic disease, but those with a family history have a higher risk. It is seen in high frequency in parts of Africa, the Mediterranean region, the Middle East, and Asia. It is also found in malaria-endemic areas."
Different types of thalassemia
Thalassemia is mainly classified into two types: Alpha-thalassemia and Beta-thalassemia. "The main difference between these two is the involvement of alpha chain and beta chain production (of hemoglobin) respectively and clinical presentation. The thalassemia you have depends on whether your alpha or beta chain contains the genetic defect," elucidated Dr Jain.
© Provided by The Indian Express thalassemia Thalassemia is an inherited blood disorder, meaning at least one of the parents must be a carrier of the disorder. (Source: Freepik)
Alpha thalassemia
In alpha thalassemia, the haemoglobin does not produce enough alpha protein. The severity depends on how many genes are mutated.
*One mutated gene: Alpha thalassemia minima is when the person has no symptoms. It happens when a healthy person who has a child with symptoms of thalassemia is a carrier.
*Two mutated genes: Alpha thalassemia minor is when person has mild anemia.
*Three mutated genes: Hemoglobin H disease is a type of chronic anemia.
*Four mutated genes: Alpha thalassemia major is the most severe form of alpha thalassemia.
Beta thalassemia
A person needs two globin genes to make beta-globin chains -- one from each parent.
*Beta thalassemia minor (beta thalassemia trait) involves having one missing or defective beta-globin gene. Some people with beta thalassemia minor don't have symptoms at all.
*Beta thalassemia intermedia may cause mild to moderate anemia symptoms. It involves having two missing or defective beta-globin genes.
*Beta thalassemia major (Cooley's anemia) is the most severe kind of beta thalassemia. It involves having two missing or defective beta-globin genes. It is also known as "transfusion-dependent thalassemia" because people with this condition require lifelong blood transfusions.
Depending on the type of thalassemia, constant medical care may be necessary to manage the condition effectively. "It can be treated through transfusion support, folate supplements, bone marrow transplant, chelating agents for iron overload management, nutrition support and genetic counselling and screening of the family members," concluded Dr Jain.
📣 For more lifestyle news, follow us on InstagramTwitterFacebook and don't miss out on the latest updates!
Genetic Mutation
Denissenko, M. F., et al. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science 274, 430–432 (1996)
Greenblatt, M. S., et al. Mutations in the P53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Research 54, 4855–4878 (1994)
International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001) (link to article)
Kimchi-Sarfaty, C., et al. "Silent" polymorphism in the MDR1 gene changes substrate specificity. Science 315, 525–528 (2006)
Mulligan, L. M., et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 363, 458–460 (1993) (link to article)
Nells, E., et al. PMP22 Thr (118) Met: Recessive CMT1 mutation or polymorphism? Nature 15, 13–14 (1997) (link to article)
Pearson, C. E., et al. Repeat instability: Mechanisms of dynamic mutations. Nature Reviews Genetics 6, 729–742 (2005) (link to article)
Pierce, B. A. Genetics: A Conceptual Approach (Freeman, New York, 2000)
Seidl, H., et al. Ultraviolet exposure as the main initiator of P53 mutations in basal cell carcinomas from psoralen and ultraviolet A-treated patients with psoriasis. Journal of Investigative Dermatology 117, 365–370 (2001)
Twyman, R. Mutation or polymorphism? Wellcome Trust website, http://genome.Wellcome.Ac.Uk/doc_WTD020780.Html (2003)
Viguera, E., et al. Replication slippage involves DNA polymerase pausing and dissociation. EMBO Journal 20, 2587–2595 (2001)
Comments
Post a Comment