Connecting Ehler’s-Danlos Syndrome and Complex Regional Pain Syndrome

Philadelphia Health Providers And Patients Seek A New Treatment Roadmap For Sickle Cell Disease After Drug Recall
From Philly and the Pa. Suburbs to South Jersey and Delaware, what would you like WHYY News to cover? Let us know!
About two weeks ago, Ediomi Utuk-Lowery got an unexpected email. It was about a medication called voxelotor, which she has been taking to help manage her sickle cell disease, an inherited blood disorder.
"I read the email, I was like, wait a minute, this can't be real," Utuk-Lowery said. "So I Googled it, and then I saw the articles, and I'm just like, no, this cannot be happening."
The email explained that Pfizer, which manufactured and sold voxelotor under the brand name Oxbryta, voluntarily pulled the medication from shelves over the company's safety concerns in ongoing clinical trials outside the United States.
The treatment drug has been on the market here since 2019, when it won approval from the Food and Drug Administration. Utuk-Lowery said it's been working well for her, increasing hemoglobin and oxygen levels in her red blood cells, which makes the recall especially devastating.
"It took a couple days for me to even be able to understand what my doctor was suggesting my weaning protocol should be, because everybody was busy," she said. "It was a shock to the community. Nobody was prepped."
Philadelphia health providers and patients say the abrupt move has left them scrambling to find alternative plans for what is a rare disorder that already has few treatment options.
Now, they are banding together as a community to find a new way forward for people living with sickle cell disease, and to increase awareness among the general public about the need for more investment in research and therapies for this disease.
"Our community definitely feels the rub with this one, and it's so important that we don't lose the long-term goal of a cure, right?" said Utuk-Lowery, who is also the co-founder of the Crescent Foundation in Philly. "This is the opportunity where you either stay focused or you get off track, and I want our community to stay focused."
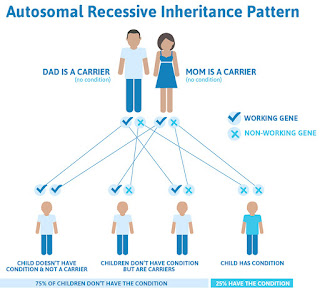
People born with sickle cell inherit the genetic condition from both parents, who either have the disease themselves or carry the genetic trait. There are four subtypes of the disease, each with varying levels of severity and health complications.
An estimated 100,000 people in the U.S. Are living with sickle cell, according to the Centers for Disease Control and Prevention. More than 90% of patients are Black or of African descent. Other patients are typically from Central and South America, or are of Middle Eastern, Asian, Indian, and Mediterranean descent.
The disease affects red blood cells, which contain the protein hemoglobin, which carries oxygen to the body's organs and tissue. Sickle cell disease impairs red blood cells' ability to hold onto oxygen, causing anemia and organ damage. Blood cells can also become rigid and warped into the shape of the letter C, so when they travel through small blood vessels, they can get painfully stuck.
"And this pain, when you talk to patients, is nearly indescribable," said Dr. Alexis Thompson, chief of the Division of Hematology at Children's Hospital of Philadelphia. "And unfortunately also, even with new treatments, it is still a condition that is associated with a shortened lifespan."
Stem cell transplants can cure the disease, but the high-risk procedure is performed on few patients and has several short- and long-term health consequences. Newer gene therapies also hold hope for long-term benefits, but they are not yet broadly accessible.
Most treatments for sickle cell manage the everyday complications of the disease, like anemia and pain. Voxelotor was approved because it showed that it could improve hemoglobin and oxygen levels in red blood cells.
It was often prescribed alongside hydroxyurea, a first-line medication used for a majority of patients. Experts say about 5 to 10% of patients in local treatment programs were taking voxelotor, which comes in a daily oral pill.
Following Pfizer's announcement on Sept. 25 to pull the drug from pharmacy and store shelves, health providers in the area quickly moved to notify patients and families of the change.
"We do not want families to be left with any more uncertainty than they have already," Thompson said. "We don't have as much information as we hope to obtain in the coming days to weeks, but the recommendation clearly is to discontinue this medication now."
There is no exact drug replacement for voxelotor, said Dr. Sophie Lanzkron, director of the Division of Hematology at Thomas Jefferson University's Sidney Kimmel Medical College. That leaves patients relying on blood transfusions or one of three other medications approved to treat the disease.
Lanzkron said some of these patients have already exhausted those other options in the past, with limited improvements.
"There are some people on voxelotor in which finding an alternative is going to be really, really hard and that breaks my heart," she said. "Like, what are we going to offer these patients?"
Without an exact roadmap for moving forward after the drug recall, Lanzkron said treatment providers in the area are collaborating to draft action plans and treatment guidance as they seek more answers from Pfizer about voxelotor.
Meanwhile, providers and patient advocates point out that if there were more treatment options to begin with, then people would not struggle so much when one medication leaves the market.
Sickle cell disease was first identified in the early 1900s, but most treatment medications were developed only in the last 30 years.
"I think it speaks to how important this is to do right by our sickle cell community and continue to understand how sickle cell works and what the next best steps are for developing new and innovative therapies that are both safe and effective," Dr. Scott Peslak, gene therapy lead at Penn Medicine's Comprehensive Sickle Cell Disease Program and Comprehensive Adult Thalassemia Program.
Peslak said the overall goal is always to keep patients safe while providing care, even if it means recalling a drug that has been on the market for several years now to investigate safety concerns and prevent potential harm.
While many patients may view this as a setback and may be feeling disappointment and anger, Utuk-Lowery said she still has high hopes for the future and encourages people to advocate for themselves and their loved ones living with this disease.
Both she and her sister live with sickle cell disease and have been participating in clinical trials for new treatments since they were kids.
"I think it's so important to remember that living with sickle cell disease should be a badge of honor. We are heroes, we are warriors," she said. "So, keep your head up, keep shining. And keep ensuring that you stand up for yourself and your community to the best of your ability."
Providers advise that patients who are or were taking voxelotor should reach out to their health care providers immediately if they have not already been contacted about the medication's recall.
WHYY is your source for fact-based, in-depth journalism and information. As a nonprofit organization, we rely on financial support from readers like you. Please give today.
Comprehensive Guide To Sickle Cell Disease Symptoms And Treatment
According to Dr. Richard Steingart, a hematologist with Baystate Hematology Oncology, roughly 100,000 Americans live with an inherited blood disorder known as sickle cell disease (SCD). Often referred to as sickle cell anemia, SCD is progressive and incurable. It can cause periods of extreme pain throughout the body and contributes to a shortened lifespan for those affected by it. But, as Dr. Steingart notes, there are promising treatment options for successfully managing SCD.
What is Sickle Cell Disease?Sickle cell disease occurs when a person inherits abnormal hemoglobin genes from both parents. Hemoglobin is a protein in red blood cells that carries oxygen throughout the body. In SCD, the abnormal hemoglobin causes normally round, flexible, red blood cells to become crescent or "sickle" shaped. In addition, these sickle-shaped cells are stiffer and stickier than normal red blood cells.
As a result, they can block the flow of blood in small blood vessels. Starved of oxygen, the tissues and organs beyond the blockage become damaged, leading to sudden and severe pain referred to as pain crises. A pain crisis can last several hours or several days. Very often, a person in pain crisis needs to go to the hospital for treatment.
In addition, sickled cells are also very fragile. While a normal red blood cell lives about 120 days, the lifespan of a sickled cell is 10 to 20 days. This high die-off rate frequently leads to anemia, which can cause an individual to tire easily and feel sluggish.
The abnormal hemoglobin causes normally round, flexible, red blood cells to become crescent or 'sickle' shaped.
Dr. Richard Steingart
What Causes Sickle Cell Disease?As Dr. Steingart explains, there are three types of hemoglobin:
"People who inherit a hemoglobin S gene from one parent and a hemoglobin A gene from the other are considered to have a sickle cell trait," says Dr. Steingart. "In most cases, these individuals usually don't have medical issues or experience pain crises. However, because they carry the S gene, they can pass it on to their children."
He adds, "Now, in instances where both parents have hemoglobin S, their children will be born with sickle cell disease versus the less-concerning sickle cell trait."
While SCD is most common in people of African descent (1 in 12), it's also found in individuals of Hispanic, Indian, European, and Mediterranean descent and exists in most countries.
How is Sickle Cell Disease Diagnosed?In the United States, every baby born in a hospital is tested for sickle cell shortly after birth. The results are shared with the family and the child's doctor, typically one to two weeks after testing.
Early detection is important as infants and children with SCD frequently suffer from more infections. As a child with SCD grows, they may also experience:
From head to toe, there's really no part of the body that can fully escape the effects of sickle cell disease.
Dr. Richard Steingart
How Does Sickle Cell Disease Affect the Body?As Dr. Steingart notes, "From head to toe, there's really no part of the body that can fully escape the effects of SCD. As mentioned, a crisis can happen at any time. Patients often describe it as a feeling of intense pressure or being stabbed by multiple knives. But even when you're not experiencing a crisis, SCD can be causing damage throughout the body."
Here a just a few of most common the impacts Dr. Steingart regularly sees:
The aim of SCD management is avoiding crises, pain relief, and preventing complications.
Dr. Steingart notes that crises are frequently brought on by the following, all of which can lead to the narrowing of blood vessels:
By abstaining from these behaviors and keeping the body warm, patients can reduce their risk of a crisis.
When crises do occur, pain management is typically accomplished with medication.
"In the best cases, over-the-counter drugs, like Tylenol, can provide the needed relief," Dr. Steingart says, "But, for severe crises, narcotics are often required."
And because people with SCD are more prone to infections, daily antibiotic treatment, often penicillin, is often recommended. In addition, routine vaccinations, including annual flu shots, are strongly encouraged.
In terms of treatment, Dr. Steingart notes that patients of today have more options and hope than ever. "For decades, there were very few options for SCD patients other than hydration and pain medication. But beginning in the late 80s and through to today, some advances have been made that significantly improve quality of life and even work to cure the disease."
Current sickle cell disease treatment options include:
HydroxyureaA cornerstone of SCD treatment since 1988, hydroxyurea works by increasing the production of hemoglobin F, which helps prevent the formation of sickled red blood cells. Hydroxyurea has been shown to reduce the frequency of pain crises, acute chest syndrome, and the need for blood transfusions.
Blood TransfusionsRegular blood transfusions are used to:
Recently, several new medications have been approved for SCD treatment:
Stem Cell or Bone Marrow Transplantation: As promising as transplantation is as potential cure for SCD, it's also fraught with risk and challenges. The first challenge is finding a suitable donor. The best option is a family member but even so, only 10-15% of family members will be a match.
In the fortunate cases where a match is found, patients must also be prepared to undergo chemotherapy, which works to wipe your system clean of "faulty" red blood cells. However, it also works to reduce immunity, meaning patients are more susceptible to illness and infection, and it also renders patients infertile.
Gene Therapy: Recently, some exciting developments in gene therapy have emerged as an option for treating SCD. Unlike transplants, gene therapy uses only the patient's blood, eliminating the obstacle of finding a suitable match.
As Dr. Steingart explains, "Gene therapy involves modifying a patient's own stem cells to produce healthy hemoglobin." Here's what's involved:
Stem Cell Collection: The patient's blood stem cells are collected from their bone marrow or blood and taken to a lab for modification.
Genetic Modification Options:
Dr. Steingart adds, "Both options require the patient to undergo chemotherapy prior to being infused with the modified genes but both aim to provide a long-lasting or potentially curative treatment by addressing the cause of SCD at the genetic level. At this point, research shows gene therapy has an encouraging success rate of 90%."
He adds, "As research continues, there is hope for even more effective treatments and potential cures in the future."
What To Know About Sickle Cell Disease (SCD)
Sickle cell disease (SCD) is an inherited health condition that slows the flow of red blood cells in your body. For people with SCD, hemoglobin S (a mutated protein that transports oxygen) causes red blood cells to become curved (sickle-shaped) and stiff. This disrupts how easily red blood cells and the oxygen they carry can move through your blood vessels.
In SCD, sickle-shaped cells clump together and block small blood vessels, leading to organ damage and other health complications. Common symptoms include severe pain, fatigue, and frequent infections. There is no cure for sickle cell disease, but there are treatment options that can reduce complications.
This type of blood disorder disproportionally affects people of African descent. In the United States, about one in 365 African American children are born with SCD. People of Hispanic, Middle Eastern, and Indian descent also have higher rates of SCD.
The type of SCD you have depends on which genes you inherited from one or both of your parents. Hemoglobin SS (HbSS): Also known as sickle cell anemia, this type of SCD comes from inheriting the hemoglobin S gene (or sickle cell gene) from both parents. Hemoglobin SC (HbSC): This type involves having two different mutated hemoglobin genes, where one parent passes on the hemoglobin S gene and the other passes on the hemoglobin C gene (a type of mutated hemoglobin). HbSC is usually less severe than HbSS. HbS beta thalassemia: This type combines hemoglobin S with another blood condition called beta thalassemia. HbS comes in two forms—beta zero and beta plus. In beta zero, the body does not make healthy hemoglobin, which leads to more serious complications. Beta plus allows some healthy hemoglobin production, so symptoms are usually milder. HbSS is the most common and severe form of sickle cell disease. Rarer types of sickle cell disease include HbSD, HbSE, and HbSO. They occur when a person inherits the hemoglobin S gene from one parent and a gene for another unusual type of hemoglobin, such as D, E, or O, from the other parent. SCD symptoms vary depending on age, the type of SCD you have, and the frequency of treatment. Symptoms of SCD may be noticeable in infants as young as five to six months old. Still, symptoms aren't the same for everyone. Some people might experience severe symptoms requiring frequent hospitalizations, while others might have milder symptoms. Sickle cell disease symptoms may include: Pain in your joints, bones, and abdomen Fatigue Anemia (low red blood cell count) Yellowing of the skin or eyes, known as jaundice Swollen hands and feet (dactylitis) Priapism (persistent and often painful erections) Frequent infections as a result of damaged spleen Delayed growth in children Sickle cell disease is an inherited condition typically passed down from parents to their children through genes. To have sickle cell disease, a person must be born with two sickle cell genes. The severity of the disease depends on how it is inherited. Getting the trait from both parents ensures you will have sickle cell disease. However, if one parent passes down the hemoglobin S gene and the other does not, the child will only carry the sickle cell trait. Although carrying sickle cell trait does not mean you have sickle cell disease, you could still pass it on to your children. Risk Factors Sickle cell disease can affect anyone, but people of African, Hispanic, and Middle Eastern descent have the highest risk of developing sickle cell disease. Sickle cell trait is also common in people from parts of Asia and India. Having two parents with the sickle cell trait increases your risk of having some form of SCD. Sickle cell disease is generally diagnosed through a blood test. In the United States, newborns are routinely screened for SCD. To do this test, a healthcare provider takes a tiny drop of blood from the baby's heel. They send the blood to a lab to check for sickle cell trait and other potential preexisting conditions. They may perform another blood test when the baby is 3-6 months old. If you have symptoms or a family history of SCD, you can request that your primary healthcare provider perform a blood test to check for sickle-shaped cells. They may also use genetic testing to look for the specific gene that causes the condition. These tests can determine whether you have the disease or the trait and help identify the type of SCD. Although there's currently no widely available cure for sickle cell disease, a hematologist can help ease symptoms through different treatment options. A bone marrow transplant is a cure that works for some people but is not as accessible for everyone. Some healthcare providers may use a red blood cell apheresis exchange to get rid of harmful red blood cells using a machine. SCD is a condition that often becomes more severe over time. The main treatment goals are alleviating pain, reducing blood cells containing HbSS, minimizing organ damage, and preventing complications. Medications A few medications can help treat different aspects of sickle cell disease. For example, hydroxyurea (Hydrea) is a commonly used treatment that can help your body make more healthy red blood cells and reduce bouts of severe pain. Other U.S. Food and Drug Administration (FDA) approved medications to treat sickle cell disease include Oxbryta (voxelotor), Endari (L-glutamine), and Adakveo (crizanlizumab). Children with SCD may be prescribed antibiotics such as penicillin to reduce their risk of infections. Pain relievers like Tylenol (acetaminophen) and Advil (ibuprofen) can help manage mild to moderate pain. A healthcare provider may prescribe stronger prescription medicines if you are experiencing severe pain. Blood Transfusions This treatment involves receiving donor blood to increase the amount of healthy red blood cells in your body. These healthy cells can carry oxygen more effectively, which helps prevent severe anemia. Blood transfusions can also lower the percentage of sickle-shaped cells in the blood, reducing the risk of other health complications like stroke. Bone Marrow Transplant This procedure replaces damaged bone marrow—the spongy tissue inside bones where blood cells are made—with healthy marrow from a donor. It helps your body produce healthy red blood cells. However, the process of receiving a transplant can be long and complicated. It requires a well-matched donor (often a family member) and involves risks such as your body not reacting well to the transplant or infection. Gene Therapy In December 2023, the FDA approved the first gene therapy to treat sickle cell disease. This treatment involves taking a person's blood or bone marrow to a lab and altering red blood cells. The modified cells are then put back into the person's body. While this is an exciting breakthrough, it's still very new, and access is limited. Since sickle cell disease is inherited, you cannot prevent yourself from getting it. If you're planning on becoming a parent, you and your partner can choose to take a genetic test to see if you are carriers of any mutated genes that can lead to SCD. This can help determine the chances that you may pass on the condition to your child or have a child who's a carrier. An internal medicine doctor or hematologist (a doctor specializing in blood disorders) can determine whether or not your child is likely to develop SCD even before birth. This involves genetic tests like examining the amniotic fluid (the fluid surrounding the fetus in the womb) or a small placenta sample. This test can be performed as early as eight to 10 weeks into the pregnancy. While there is not yet a cure for sickle cell disease, there are ways to reduce the severity of some symptoms. Steps you can take to alleviate pain, reduce infections, and the risk of other health conditions include the following: Stay hydrated Be mindful of hydration and protection in extreme heat or cold Keep up with vaccinations Follow a nutritious eating plan Exercise regularly Take medications as recommended by your healthcare provider Visit your hematologist and other specialists on your healthcare team for regular screenings Avoid high altitude environments (flying, mountain climbing, and more) when possible As sickle-shaped cells move through your bloodstream, they can damage organs and tissues in your body. Organ and tissue damage from SCD can cause serious complications, including: Acute chest syndrome: This is a severe lung complication where sickle cells block blood flow in the lungs, causing chest pain and trouble breathing. Avascular necrosis: When this occurs, bone tissue dies due to a lack of blood supply. It most often affects the hip joint. Blood clots: Sickle cells can cause blood to clot more quickly, blocking blood flow to different parts of your body. This can lead to deep vein thrombosis (DVT), when blood clots form in the large veins, usually in your legs. Kidney problems: Sickle cells can limit blood flow to your kidneys. This can lead to kidney damage or chronic kidney disease (CKD), making it difficult for your kidneys to clean your blood effectively. Liver issues: Your liver may become damaged from working overtime to replace sickled cells. Pulmonary hypertension: High blood pressure can occur due to blood vessel damage, making it hard for your heart to pump blood through your lungs. Pulmonary hypertension is a potentially life-threatening condition. Splenic sequestration: Sickle cells can get trapped in the spleen, causing it to enlarge. Stroke: Sickle cells block blood vessels and cut off oxygen to your brain. Vision loss: Sickle cells can damage blood vessels in the eye, leading to vision problems or blindness. Sickle cell disease is a blood disorder where the altered protein hemoglobin S changes the shape of red blood cells into a sickle shape. This makes it more difficult for red blood cells to move throughout your body. This disruption in blood flow can cause intense pain, anemia, fatigue, and shortness of breath, among other symptoms. More severe cases may result in organ damage and blurry vision. Every person who carries the sickle cell trait will not automatically develop SCD. However, if both of your parents carry a gene for SCD, you are more likely to develop the disease. A hematologist can help you come up with a treatment plan for living with SCD. Although there is no widely available cure for SCD, there are various treatments and medications available to help manage symptoms.Thanks for your feedback!


Comments
Post a Comment