Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement From the American Heart Association

Mendelian Genetics: Patterns Of Inheritance And Single-Gene Disorders
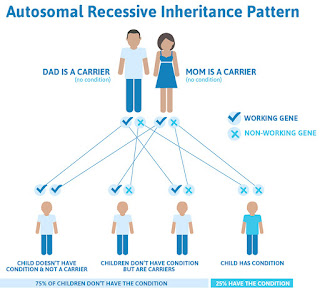
Autosomal recessive single-gene diseases occur only in individuals with two mutant alleles of the disease-associated gene. Remember, for any given gene, a person inherits one allele from his or her mother and one allele from his or her father. Therefore, individuals with an autosomal recessive single-gene disease inherit one mutant allele of the disease-associated gene from each of their parents. In pedigrees of families with multiple affected generations, autosomal recessive single-gene diseases often show a clear pattern in which the disease "skips" one or more generations.
Phenylketonuria (PKU) is a prominent example of a single-gene disease with an autosomal recessive inheritance pattern. PKU is associated with mutations in the gene that encodes the enzyme phenylalanine hydroxylase (PAH); when a person has these mutations, he or she cannot properly manufacture PAH, so he or she is subsequently unable to break down the amino acid phenylalanine, which is an essential building block of dietary proteins. As a result, individuals with PKU accumulate high levels of phenylalanine in their urine and blood, and this buildup eventually causes mental retardation and behavioral abnormalities.
The PKU-associated enzyme deficiency was determined biochemically in the 1950s—long before the PAH-encoding gene was mapped to human chromosome 12 and cloned in 1983. Specifically, Dr. Willard Centerwall, whose child was mentally handicapped, developed the first diagnostic test for PKU in 1957. Called the "wet diaper" test, Centerwall's test involved adding a drop of ferric chloride to a wet diaper; if the diaper turned green, the infant was diagnosed with PKU. The wet diaper test was used to reliably test infants at eight weeks after birth; by this time, however, infants who were affected by PKU had already often suffered irreversible brain damage.
Thus, in 1960, Dr. Robert Guthrie, whose niece suffered from PKU and whose son was also mentally handicapped, established a more sensitive method for detecting elevated phenylalanine levels in blood, which permitted a diagnosis of PKU within three days after birth. Guthrie's test used bacteria that were unable to make their own phenylalanine as messengers to report high blood levels of phenylalanine in an infant's blood sample obtained via heel prick. With Guthrie's method, the phenylalanine-deficient bacteria were grown in media together with a paper disk spotted with a drop of the infant's blood. If the phenylalanine levels in the blood were high, the bacteria would grow robustly, and a diagnosis of PKU could be made. Through the ability to discover that their child had PKU at such an early age, parents became able to respond immediately by feeding their child a modified diet low in proteins and phenylalanine, thereby allowing more normal cognitive development. Guthrie's test continues to be used today, and the practice of obtaining an infant's blood sample via heel prick is now used in numerous additional diagnostic tests.
Several other human diseases, including cystic fibrosis, sickle-cell anemia, and oculocutaneous albinism, also exhibit an autosomal recessive inheritance pattern. Cystic fibrosis is associated with recessive mutations in the CFTR gene, whereas sickle-cell anemia is associated with recessive mutations in the beta hemoglobin (HBB) gene. Interestingly, although individuals homozygous for the mutant HBB gene suffer from sickle-cell anemia, heterozygous carriers are resistant to malaria. This fact explains the higher frequency of sickle-cell anemia in today's African Americans, who are descendants of a group that had an advantage against endemic malaria if they carried HBB mutations. Finally, oculocutaneous albinism is associated with autosomal recessive mutations in the OCA2 gene. This gene is involved in biosynthesis of the pigment melanin, which gives color to a person's hair, skin, and eyes.
Little-studied RNA Might Be Key To Regulating Genetic Disorders Like Epilepsy And Autism
Just a moment...This request seems a bit unusual, so we need to confirm that you're human. Please press and hold the button until it turns completely green. Thank you for your cooperation!
Press and HoldPress and hold the button
If you believe this is an error, please contact our support team.
167.71.87.121 : cc69e672-6e34-406e-a207-71e8d124
Gene Mutation May Explain Rare Neurological Disorders
Register for free to listen to this article
Thank you. Listen to this article using the player above. ✖
Researchers identified rare genetic mutations that may account for undiagnosed neurodevelopmental disorders, linking the FLVCR1 gene with various neurological symptoms.
Investigating a genetic anomalyA collaborative study, led by Baylor College of Medicine and the National University of Singapore, has found a genetic diagnosis for 30 individuals who had undiagnosed neurodevelopmental disorders despite extensive testing. The study focused on a rare mutation in the FLVCR1 gene, known to play a role in red blood cell production and the cellular transport of choline and ethanolamine. Prior evidence indicated that these molecules are critical for maintaining cell membrane integrity and supporting cell division.
FLVCR1 gene FLVCR1 encodes a protein involved in cellular transport of choline and ethanolamine, molecules essential for cell membrane stability and function. This gene is critical in producing red blood cells and may influence neurodevelopment. Choline Choline is an essential nutrient involved in cell membrane structure and nerve function. It's a precursor for acetylcholine, a neurotransmitter, and phosphatidylcholine, a component of cell membranes. Choline deficiencies can lead to neurodevelopmental issues, liver disease, and anemia. Ethanolamine Ethanolamine is a compound necessary for forming phospholipids like phosphatidylethanolamine, which help maintain cell membrane integrity and assist in various cellular processes. It plays a role in nervous system function and neurodevelopment.
Initial investigations were prompted by one patient with a severe neurodevelopmental disorder, epilepsy and an unusual lack of pain sensitivity. Despite extensive genetic tests, clinicians could not determine a cause. Enrolling in the Baylor GREGoR (Genomics Research to Elucidate the Genetics of Rare Diseases) research program enabled reanalysis, which pointed to the FLVCR1 gene as a potential source of these symptoms.
Want more breaking news?Subscribe to Technology Networks' daily newsletter, delivering breaking science news straight to your inbox every day.
Subscribe for FREE The role of FLVCR1 in neurodevelopmental disordersWhile some previous studies on the FLVCR1 gene in animals suggested it might contribute to bone malformations and red blood cell issues, it had not been definitively linked to similar human conditions. For instance, mice with Flvcr1 gene knockouts had severe bone and blood cell development issues that paralleled symptoms of Diamond-Blackfan anemia (DBA) in humans. However, DBA has typically been associated with other genes.
FLVCR1 mutations have also been observed in people with ataxia – a neurodegenerative disorder marked by poor muscle control and coordination – accompanied by sensory and vision impairments. This pattern differed from the severe developmental conditions seen in the new patient cases, prompting researchers to question if FLVCR1 mutations might contribute to a broader spectrum of neurological symptoms.
"We were intrigued. On one hand, we had a patient with a rare FLVCR1 mutation and severe developmental conditions, epilepsy and complete insensitivity to pain, but on the other hand there were patients with rare mutations on the same gene that presented with a different set of problems. Could it be that those different mutations of FLVCR1 caused not one set but a spectrum of characteristics we observed in all the patients combined?"
Dr. Daniel Calame
Expanding the patient databaseTo investigate this hypothesis, researchers analyzed large datasets of individuals with undiagnosed neurodevelopmental conditions and identified others with FLVCR1 gene mutations. Through databases such as the Baylor-Hopkins Center for Mendelian Genomics, GeneMatcher, and other clinical sources, they located 30 patients across 23 families with similar symptoms. Within this group, researchers identified 22 unique FLVCR1 gene variants, including 20 that had not been previously documented.
These individuals showed overlapping symptoms with the mouse models, including anemia, brain malformations, and bone deformities. These features suggested FLVCR1 may play a broader role in both human and animal neurodevelopmental disorders than previously understood.
Testing gene function in the laboratoryThe research team, in collaboration with the Yoon Long Lin School of Medicine, assessed how different FLVCR1 gene mutations impact choline and ethanolamine transport in cells. Lab testing revealed that these gene variants reduced the transport efficiency of choline and ethanolamine by as much as 50%. This finding supports prior studies indicating that choline is crucial for normal neurodevelopment. Deficiencies in choline have also been associated with anemia, growth delays and immune deficiencies, conditions observed in patients with FLVCR1 mutations.
The results suggest that the severity of neurodevelopmental symptoms may correlate with the degree to which a FLVCR1 variant affects choline transport. This mechanistic insight provides a clearer understanding of how FLVCR1 mutations could result in a spectrum of symptoms from multiorgan developmental disorders to adult-onset neurodegeneration.
Potential implications for diagnosis and treatmentThe study highlights the importance of examining multiple data sources when analyzing rare genetic conditions, particularly for genes like FLVCR1 with wide-ranging effects. In these cases, consulting animal models proved valuable, as the mouse studies pointed to similarities with DBA-like symptoms that were not initially linked to FLVCR1 in humans. This broader approach allowed researchers to identify the FLVCR1 gene variants as the likely cause for several long-standing undiagnosed conditions.
"The 30 patients we identified had not had a diagnosis for years; it was rewarding to be able to provide an explanation for their condition."
Dr. Daniel Calame
Furthermore, the findings suggest that choline or ethanolamine supplementation may have potential as a treatment for individuals with FLVCR1-related neurodevelopmental disorders, though further research is needed. For now, the study's contribution lies in providing a diagnosis for individuals whose conditions had remained a mystery for years.
Reference: Calame DG, Wong JH, Panda P, et al. Biallelic variation in the choline and ethanolamine transporter FLVCR1 underlies a severe developmental disorder spectrum. Genetics Med. 2024:101273. Doi: 10.1016/j.Gim.2024.101273
This article has been republished from the following materials. Note: material may have been edited for length and content. For further information, please contact the cited source. Our press release publishing policy can be accessed here.
This content includes text that has been generated with the assistance of AI. Technology Networks' AI policy can be found here.


Comments
Post a Comment